
飞秒时间分辨实验中泵浦-探测交叉相关函数的测量和时间零点的确定
2017-06-01 11:29布玛丽亚阿布力米提
发光学报 2017年5期
布玛丽亚·阿布力米提, 向 梅
(1. 新疆师范大学 物理与电子工程学院, 新疆 乌鲁木齐 830054;2. 中国科学院武汉物理与数学研究所 波谱与原子分子物理国家重点实验室, 湖北 武汉 430071)
飞秒时间分辨实验中泵浦-探测交叉相关函数的测量和时间零点的确定
布玛丽亚·阿布力米提1,2*, 向 梅1
(1. 新疆师范大学 物理与电子工程学院, 新疆 乌鲁木齐 830054;2. 中国科学院武汉物理与数学研究所 波谱与原子分子物理国家重点实验室, 湖北 武汉 430071)
飞秒激光技术的出现使得实时探测与跟踪激发态超快弛豫动力学过程成为可能,并能够给出激发态动力学过程清晰的物理图像。而在飞秒时间分辨实验中,泵浦-探测相关函数和时间零点直接影响实验结果的可靠性和准确性。本文结合飞秒激光在分子激发态超快动力学过程中的应用进展,介绍了根据实验条件和要求,在具体实验过程中泵浦-探测相关函数测量和时间零点确定的几种方法。实验中选择可见光作为泵浦光和探测光时,可以通过测定随泵浦-探测时间延迟变化的泵浦激光与探测激光的和频/差频光强来确定泵浦探测交叉相关函数和时间零点;而选择中心波长在紫外甚至真空紫外的激光脉冲作为泵浦光或探测光时,泵浦-探测交叉相关函数通常采用校正的方法测量。
飞秒激光; 泵浦-探测; 相关函数; 时间分辨
1 引 言
稳定的分子通常处于分子能量最低的能态,即基态。能量比基态高但尚未电离的状态为激发态。原子或分子吸收一定的能量以后,从基态跃迁到激发态。激发态被布居以后,由于激发态的不稳定性,很容易通过各种衰减通道进行衰减。在超短脉冲激光出现以前,人们常通过一些间接的手段研究激发态的衰减过程并给出有关激发态的信息,如测量自然线宽的方法可估算激发态的寿命[1]。通过自然线宽来测量激发态的寿命时,由于做一些理想的假设并考虑到众多因素,在实际的实验测量中很难实现。但这种方法的优点是在大多数情况下可以很容易获得激发态寿命的下限。除了从自然线宽中得到激发态寿命的下限以外,通过一些其他的间接方法也能够估算激发态寿命的下限,如产物角分布、吸收线宽、拉曼极化等,但时间分辨测量更直接和明确。飞秒激光技术[2]的出现实现了对激发态寿命的直接测量[3]、激发态动力学的实时探测[4-5]、过渡态的实时跟踪[6],并能够给出相关动力学过程的清晰的物理图像。
在飞秒时间分辨的实验中,通常将泵浦-探测技术与光谱技术相结合。将一束飞秒光作为泵浦光使分子激发到激发态上,用另外一束光在不同泵浦-探测时间延迟下把激发态上的分子激发到实验上可观测态上并探测该分子不同时刻的光谱,从而可以测得激发态的准确寿命,也可在飞秒时间尺度上实时探测分子激发态超快动力学过程。
2 相关理论
在飞秒时间分辨实验中通常利用两束飞秒光分别作为泵浦光和探测光,这两束飞秒激光脉冲都是具有一定脉冲宽度的高斯脉冲。除此之外,实验中用到的光电倍增管等光电转换器件都不可能瞬间响应,都有一定的仪器响应时间。因此,在飞秒泵浦-探测实验中所记录的在不同时刻的离子/电子信号强度I(t)包括激发态布居数随时间变化的真实信号[X]和泵浦-探测交叉相关函数G(t)的卷积[7],即:
(1)如果假设分子吸收一个或多个光子激发到激发态A, 由于激发态不稳定,被光激发的激发态A会衰减,A态衰减到另外一个激发态B,B态也有可能衰减到C态,第一步的速度常数为k1,第二步的速度常数为k2,这样一个(A→B→C)反应过程的速率方程如下:
(2)
(3)
(4)
(5)由此可见,激发态布居数随时间的变化为一个单指数衰减、单指数上升或单指数衰减加单指数上升函数。由于两个高斯脉冲的相关函数仍然是一个高斯函数,飞秒泵浦光和探测光的相关函数G(t)为高斯函数e-a2t2。因此,对A、B、C态的布居数随时间的变化进行测量时,在实验中所记录的信号为

(6)
(7)
(8)
式中t为泵浦-探测延迟时间。由此可见,实验直接记录的信号强度随泵浦-探测延迟时间变化的信号I(t)反映的并不仅是激发态随时间演变信号,而是激发态随时间演变信号和泵浦-探测交叉相关函数的卷积。因此,在飞秒时间分辨实验中先测量泵浦-探测交叉相关函数G(t),然后对信号I(t) 进行去卷积处理,才能获得研究体系激发态随时间演变信号 [X](X=A,B,C)。此外,为了保证实验数据的准确性和可靠性,准确地确定零点也是非常重要的步骤。因此,在飞秒时间分辨实验中,测量泵浦-探测交叉相关函数和准确确定时间零点是一个非常重要的工作。
3 交叉相关函数的测量与零点的确定
飞秒激光[8-9]的一些参数并不是非常稳定,每次重新锁模后很多参数都有可能发生一些变化,因此,为了保证测量的实验数据的准确性和一致性,每次飞秒时间分辨实验前后必须测量泵浦-探测交叉相关函数和泵浦光、探测光功率。在实验过程中,由于每次研究的体系可能不同,经常选择不同波长的光作为泵浦光和探测光。由于不同的实验所选择的泵浦光和探测光的波长不同,同时不同实验室本身实验条件也不同,不同的实验通常选择不同的方法测量泵浦-探测交叉相关函数和确定时间零点。下面以飞秒时间分辨超快动力学的研究进展为例,介绍飞秒泵浦光-探测交叉相关函数的测量和确定时间零点的几种方法。
3.1 光学方法确定泵浦-探测交叉相关函数和时间零点
用飞秒时间分辨的泵浦-探测技术对分子激发态超快动力学过程进行研究时,泵浦光和探测光可能选用基频光(ω,中心波长800 nm)、二倍频光(2ω,中心波长400 nm)或三倍频光(3ω,中心波长约266 nm)作为泵浦光和探测光,采用共振增强多光子电离(REMPI)的方法制备激发态量子波包或者对制备的量子波包进行探测。如中国科学院武汉物理与数学研究所张冰研究员研究小组利用400 nm为泵浦光,800 nm 为探测光研究了CH3I、C2H5I[10]等分子预解离动力学过程,研究了C6H4(CH3)2[11]、BzCl[12]分子激发态的内转换非绝热动力学过程。在这类实验中,可以通过测定随泵浦-探测时间延迟而变化的泵浦激光和探测激光和频光(如:800 nm和400 nm的和频光为 266 nm,266 nm和800 nm的和频光为200 nm)或差频光(266 nm和400 nm的差频光为800 nm)的强度来确定泵浦激-探测交叉相关函数和时间零点。本实验是在中国科学院武汉物理与数学研究所波谱与原子分子物理国家重点实验室基频光输出的飞秒激光系统完成的。该基频光输出的飞秒激光系统由3个部分组成,分别为钛宝石振荡器、啁啾脉冲放大系统和泵浦激光系统[13]。自锁模钛宝石振荡器输出脉宽约为20 fs的窄脉冲种子光,种子光进入啁啾脉冲放大系统进行能量放大,得到中心波长为800 nm、单脉冲能量为1.0 mJ、工作频率为1 kHz的飞秒基频光。图1为用基频光和二倍频光和频的方法测量交叉相关函数的光路图。

图1 基频光和二倍频光和频的方法测量交叉相干函数的光路图
Fig.1 Sketch map of light line for determining the cross-correlation function and the zero of time
光路中添加了精密控制的位移平台和三倍频晶体(BBO)。实验中选用合适的BBO晶体,对基频光(ω)进行倍频得到二倍频光(2ω),基频光和二倍频光通过合适的BBO晶体和频后可以得到三倍频光(3ω)。泵浦激光和探测激光可以共线或者交叉入射的方式引入BBO晶体。采用交叉入射方式,通过BBO晶体后几个光较容易分开;而采用共线入射方式由于简化了测量光程的过程,操作起来相对容易,也便于光聚焦。本实验采用泵浦光和探测光同向共线入射的方式进入BBO晶体。利用三棱镜解决了从BBO晶体中输出后几个光的分开问题。虽然三棱镜色散会造成和频脉宽的增宽,但由于所测量的相关函数反映的是和频光强与泵浦-探测延迟时间的关系,和频光脉冲宽度的增宽并不会对测量的泵浦-探测交叉相关函数产生影响。在实验中,通过移动位移平台改变泵浦光和探测光的光程,实现泵浦-探测时间延迟。利用二极管接收不同泵浦-探测时间延迟下的和频光信号,可以测得泵浦光和探测光的相关函数。如图2为基频光(ω)和二倍频光(2ω)的交叉相关函数。
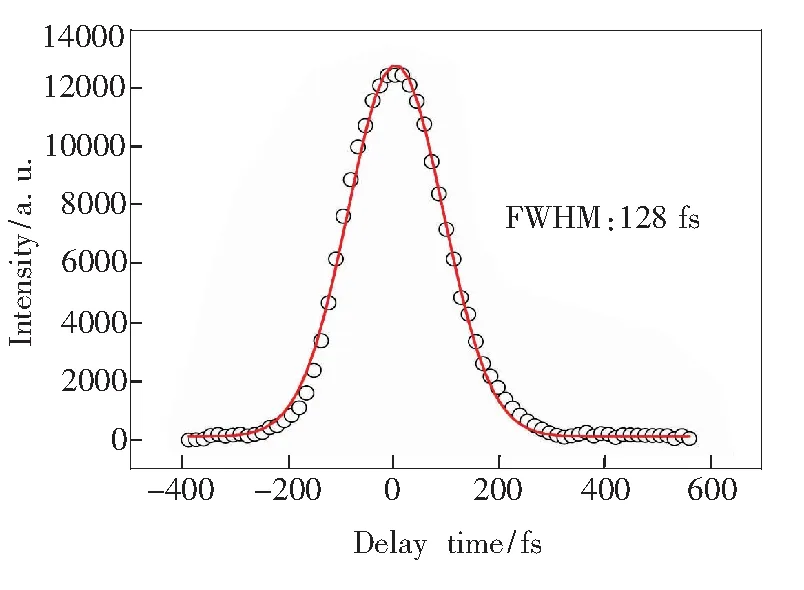
图2 泵浦-探测交叉相关函数
根据交叉相关函数的定义,交叉相关函数中信号强度最大值对应的点是泵浦光和探测光两束光时间上完全重合的等光程的点。在飞秒时间分辨测量中这一点即设为时间零点。
3.2 校正方法确定探测光的相关函数和时间零点的确定
对分子激发态和里德堡态的超快动力学过程进行研究时,泵浦光或探测光经常需要选择中心波长在紫外(UV)甚至真空紫外(VUV)的激光脉冲[14]。如张冰研究员研究小组选用266 nm作为泵浦激光,800 nm 作为探测光,研究了C6H7N[15]、CS2[16]等分子的电子激发态超快动力学过程。Suzuki教授小组研究苯的超快动力学过程时,用VUV激光探测苯分子的激发态动力学[17]。Allison等利用波长161 nm的VUV超快脉冲,在飞秒时间尺度上探测乙炔气体在强激光作用下的产物[18]。Oliver等利用272~238 nm为泵浦光,300 nm为探测光研究了苯胺与同分异构体的超快动力学过程[19]。Sonia等利用193 nm光泵浦,利用CH3和I的共振波长探测,研究了碘甲烷分子的预解离动力学过程[20]。Zewail教授小组利用UV波段的飞秒激光研究了CH3I、C2H5I、n-C3H7I和n-C4H9I等分子的超快动力学过程[21]。Suzuki教授小组把飞秒VUV光作为探测光观察到了吡嗪分子S2/S1和S1/S0之间的内转换动力学过程[22],把飞秒VUV光作为探测光研究了呋喃分子衰减寿命极短的里德堡态的动力学过程[23]。在这些实验中,泵浦激光脉冲或探测激光脉冲本身就是一次或者多次通过非线性晶体和频后获得的单脉冲能量较低的光,这时虽然理论上也可以利用前述光学的方法来确定泵浦-探测交叉相关函数,但是技术上很有难度。因此,在这些实验中,通常采用进行校正的方法测量泵浦-探测交叉相关函数。
在校正方法中,常用从长寿命分子信号中提取泵浦-探测相关函数[24]、利用某些原子[13]或分子的非共振电离信号来确定泵浦激光与探测激光的相关函数[25]、样品气体中加入微量的寿命短但电离信号强的分子作为校正分子做平行实验等方法测量泵浦光和探测光的相关函数。
实验中可以利用长寿命的分子作为校正分子,对长寿命分子离子信号进行微分,可以获得泵浦光和探测光的相关函数。如实验中采用的泵浦光是 226 nm附近的短波长的光时,可选用NO分子作为测量交叉相关函数的校正分子[26]。NO的A(2Σ+)态是寿命为几百个纳秒的长寿命的态,选择中心波长为226 nm的泵浦光可以泵浦 NO的 A(2Σ+)态的00振动模。由于对飞秒泵浦-探测交叉相关函数而言这个大于几百纳秒的寿命可以认为是无限长,因此,对在实验中测得的信号进行微分可以得到泵浦-探测交叉相关函数。图3(a)显示的是实验中测得的信号,图3(b)中的泵浦-探测交叉相关函数是高斯函数,时间尺度为290 fs。交叉相关函数的最强的点也就是泵浦探测实验的时间零点。
紫外波段的光作为泵浦光的实验中,一般也可以用氙气(Xe)等作为校正分子,利用这种原子气体的非共振电离信号来确定泵浦-探测交叉相关函数[27]。丙酮分子的能级结构比较特殊,它的第一个吸收带位于330~220 nm 波段,是源于π*←n的禁戒(Dipole-forbidden)跃迁,第一电子激发态带源位于328.6 nm。它的第二个吸收带是个很强的吸收带,位于195~180 nm 的波段范围,是源于3s←n0跃迁,第二电子激发态带源位于 195.2 nm。丙酮分子的第一电子激发态和第二电子激发态的能级差高达2.6 eV (250 kJ/mol),这种能级结构在其他分子体系中不常见。丙酮分子的这种能级特殊的结构有利于进行泵浦-探测交叉相关函数的测量。如选用的泵浦光波长是能激发丙酮至无共振能级区的激光,由于泵浦到的是丙酮分子的无共振能级区,可以把丙酮分子母体离子时间分辨强度信号作为飞秒激光泵浦-探测交叉相关函数。

图3 (a) 在实验中测得的NO在226 nm泵浦、297 nm探测下的离子信号随泵浦探测时间延迟的变化;(b) 泵浦光和探测光的交叉相关函数(290 fs)[26]。
Fig.3 (a) Time-resolved total ion signal as a function of delay time, between the pump pulse at 226 nm and the probe pulse at 297 nm. (b) Pump-probe cross-correlation function[26].
当实验中所选用激光脉冲的中心波长为更短时,由于单脉冲能量过低,很可能得不到信噪比较好的Xe 时间演化信号。因此,实验中选用VUV波段的光时,需要寻找其他方法确定泵浦-探测交叉相关函数和时间零点。目前最常用的方法是在样品气体中加入微量电离信号强而寿命很短的分子,还可以直接用一些短寿命分子做平行实验。苯、甲苯等分子被VUV波段的光激发后寿命很短,可以作为校正分子。在实验中主要是由于校正分子某些态寿命比泵浦-探测交叉相关函数还要短,因此,这些态的共振增强多光子电离时间演化信号能很近似地反映出泵浦-探测交叉相关函数。
如果时间分辨实验中的激光光源是可调谐飞秒激光,泵浦-探测交叉相关函数的测量可以不用校正分子。这时调节可调谐激光波长,激发样品分子一个已知的短寿命态,即可获得所用泵浦-探测交叉相关函数和时间零点。
在飞秒时间分辨的实验中,测得泵浦-探测交叉相关函数后,对实验上测得的信号进行去卷积处理才能得到被研究体系激发态真正的衰减函数。去卷积,数学上已经是完全可以解决的问题。但在实际测量中采集的点再多,离子信号衰减数据也是一些离散的点,对离散的点去卷积具有一定的难度。人们发展了线性和非线性最小二乘法、矩阵法、Four1er变换[28]等方法,解决了离散点去卷积时的困难。对去卷积后得到的质谱数据通常用一组函数(多数情况下是指数衰减、指数上升或指数衰减加指数上升函数)拟合,可以获得激发态的寿命参数。
4 结 论
利用飞秒激光脉冲泵浦-探测技术进行时间分辨的实验时,泵浦-探测交叉相关函数和时间零点的确定是十分关键的步骤。在不同的实验条件下,需要考虑采用不同的方法。当实验中选择可见波段的飞秒激光作为泵浦光和探测光时,可以通过测量随泵浦-探测延迟时间而变化的泵浦光和探测光二者的和频光强实现;当实验中选择紫外甚至更短的波段飞秒激光作为泵浦光和探测光时,目前还很难利用直接的技术手段来测定泵浦-探测交叉相关函数及时间零点,通常采用的做法是选择合适的原子气体或分子作为校正分子进行间接测量。
[1] 尹淑慧. 飞秒实时探测技术研究小分子里德堡态动力学 [D]. 大连: 中国科学院大连化学物理研究所, 2003. YIN S H.FemtosecondRealTimeProbeTechniquetoInvestigateTheRydbergStatesDynamicsofSmallMolecules[D]. Dalian: Dalian Institute of Chemical Physics, Chinese Academy of Sciences, 2003. (in Chinese)
[2] 张扬, 钱静, 李鹏飞, 等. 飞秒激光诱导的Mn2+掺杂锗酸盐玻璃上转换发光 [J]. 发光学报, 2015, 36(7):738-743. ZHANG Y, QIAN J, LI P F,etal.. Upconversion luminescence of Mn2+doped-germanate glass induced by femtosecond laser pulses [J].Chin.J.Lumin., 2015, 36(7):738-743. (in Chinese)
[3] NOLLER B, POISSON L, MAKSIMENKA R,etal.. Femtosecond dynamics of isolated phenylcarbenes [J].J.Am.Chem.Soc., 2008, 130(45):14908-14909.
[4] ABULIMITI B, ZHU R S, QIU X J,etal.. Studies of ultrafast dynamics of 3-picoline with femtosecond time-resolved photoelectron Imaging [J].ActaPhys.-Chim.Sinica, 2014, 30(1):22-27.
[5] LIU Z M, WANG Y M, HU C L,etal.. Photodissociation dynamics of 2-iodotoluene investigated by femtosecond time-resolved mass spectrometry [J].Chin.J.Chem.Phys., 2016, 29(1):53-58.
[6] ZEWAIL A H. Femtochemistry: atomic-scale dynamics of the chemical bond [J].J.Phys.Chem. A, 2000, 104(24):5660-5694.
[7] LI B L, MYERS A B. Emission polarization in the S3state of CS2vapor as a probe of predissociation: consideration of the finite bandwidth of the incident field [J].J.Chem.Phys., 1991, 94(4):2458-2468.
[8] 闫焱, 李凌. 飞秒激光照射金箔的分子动力学模拟 [J]. 光学学报, 2016, 36(8):0814001-1-6. YAN Y, LI L. Molecular dynamics simulation of femtosecond laser irradiating gold foils [J].ActaOpt.Sinica, 2016, 36(8):0814001-1-6. (in Chinese)
[9] 李晨, 程光华. 飞秒激光诱导金属钨表面周期性自组织结构的研究 [J]. 光学学报, 2016, 36(5):0532001-1-6. LI C, CHENG G H. Investigation of femtosecond laser induced periodic surface structure on tungsten [J].ActaOpt.Sinica, 2016, 36(5):0532001-1-6. (in Chinese)
[10] XU Y Q, QIU X J, ABULIMITI B,etal.. Energy transfer of ethyl iodine studied by time-resolved photoelectron imaging [J].Chem.Phys.Lett., 2012, 554:53-56.
[11] LIU Y Z, TANG B F, SHEN H,etal.. Probing ultrafast internal conversion of o-xyleneviafemtosecond time-resolved photoelectron imaging [J].Opt.Express, 2010, 18(6):5791-5801.
[12] DING Z H, QIU X J, XU Y Q,etal.. Ultrafast internal conversion dynamics of benzyl chloride by femtosecond time-resolved photoelectron imaging [J].ActaPhys.-Chim.Sinica, 2012, 28(12):2761-2766.
[13] LONG J Y, QIN C C, LIU Y Z,etal.. Direct imaging of the Fermi resonance interaction in para-difluorobenzene: a special insight into energy redistributions in the S1low-energy regime [J].Phys.Rev. A, 2011, 84(6):063409.
[14] LUCAS M, LIU Y L, BRYANT R,etal.. Vacuum ultraviolet photodissociation dynamics of methanol at 121.6 nm [J].Chem.Phys.Lett., 2015, 619:18-22.
[15] ABULIMITI B, ZHU R S, LONG J Y,etal.. Study on ultrafast dynamics of 2-picoline by femtosecond time-resolved photoelectron imaging [J].J.Chem.Phys., 2011, 134(23):234301-1-6.
[16] LONG J Y, LIU Y Z, QIN C C,etal.. Real-time visualization of the dynamic evolution of CS24d and 6s Rydberg wave packet components [J].Opt.Express, 2011, 19(5):4542-4552.
[17] LIU S Y, OGI Y, FUJI T,etal.. Time-resolved photoelectron imaging using a femtosecond UV laser and a VUV free-electron laser [J].Phys.Rev. A, 2010, 81(3):031403.
[18] ALLISON T K, WRIGHT T W, STOOKE A M,etal.. Femtosecond spectroscopy with vacuum ultraviolet pulse pairs [J].Opt.Lett., 2010, 35(21):3664-3666.
[19] KIRKBY O M, SALA M, BALERDI G,etal.. Comparing the electronic relaxation dynamics of aniline and d7-aniline following excitation at 272-238 nm [J].Phys.Chem.Chem.Phys., 2015, 17(25):16270-16276.
[20] POULLAIN S M, GONZLEZ M G, SAMARTZIS P C,etal.. New insights into the photodissociation of methyl iodide at 193 nm: stereodynamics and product branching ratios [J].Phys.Chem.Chem.Phys., 2015, 17(44):29958-29968.
[21] CORRALES M E, LORIOT V, BALERDI G,etal.. Structural dynamics effects on the ultrafast chemical bond cleavage of a photodissociation reaction [J].Phys.Chem.Chem.Phys., 2014, 16(19):8812-8818.
[22] HORIO T, SPESYVTSEV R, NAGASHIMA K,etal.. Full observation of ultrafast cascaded radiationless transitions from S2(ππ*) state of pyrazine using vacuum ultraviolet photoelectron imaging [J].J.Chem.Phys., 2016, 145(4):044306.
[23] SPESYVTSEV R, HORIO T, SUZUKI Y I,etal.. Excited-state dynamics of furan studied by sub-20-fs time-resolved photoelectron imaging using 159-nm pulses [J].J.Chem.Phys., 2015, 143(1):014302.
[24] SHEN H, ADACHI S, HORIO T,etal.. Two-color deep-ultraviolet 40-fs pulses based on parametric amplification at 100 kHz [J].Opt.Express, 2011, 19(23):22637-22642
[25] WU G R, NEVILLE S P, SCHALK O,etal.. Excited state non-adiabatic dynamics of pyrrole: a time-resolved photoelectron spectroscopy and quantum dynamics study [J].J.Chem.Phys., 2015, 142(7):074302.
[26] 布玛丽亚·阿布力米提. 含氮芳香烃化合物的非绝热动力学研究 [D]. 武汉: 中国科学院大学(武汉物理与数学研究所), 2013. BUMALIYA A.StudyofNonadiabaticDynamicsofNitrogen-containingAromaticCompounds[D]. Wuhan: Wuhan Institute of Physics and Mathematics (WIPM) of Chinese Academy of Sciences, 2013. (in Chinese)
[27] 尹淑慧, 刘红平, 张建阳, 等. 飞秒时间分辨质谱方法研究CF3I光电离动力学 [J]. 化学物理学报, 2003, 16(1):3-8. YIN S H, LIU H P, ZHANG J Y,etal.. Studies of photoionization of CF3I by femtosecond time-resolved mass spectrometry [J].Chin.J.Chem.Phys., 2003, 16(1):3-8. (in Chinese)
[28] BARONAVSKI A P, OWRUTSKY J C. Lifetime of the S3state of CS2measured by femtosecond ultraviolet multiphoton ionization spectroscopy [J].Chem.Phys.Lett., 1994, 221(5-6):419-425.

布玛丽亚·阿布力米提(1984-),女,新疆阿图什人,博士,副教授,2014年于中国科学院大学获得博士学位,主要从事超快动力学、超快激光等方面的研究。
E-mail: maryam917@163.com
Determine The Pump-probe Cross Correlation Function and The Zero of Time of The Pump and Probe Laser in Femtosecond Time-resolved Studies
Bumaliya·ABULIMITI1,2*, XIANG Mei1
(1.SchoolofPhysicsAndElectronicEngineering,XinjiangNormalUniversity,Urumqi830054,China;2.StateKeyLaboratoryofMagneticResonanceandAtomicandMolecularPhysics,WuhanInstituteofPhysicsandMathematics,ChineseAcademyofSciences,Wuhan430071,China)
Femtosecond laser technique, not only makes the observation of excited state relaxation processes more directly, but also offers information for disentangle the complex dynamics of excited state relaxation processes. However, what have an explosive impact on accuracy and reliability of experimental result is that cross correlation function as well as the zero of time of the pump and probe laser pulses, therefore, determine the correct value of them is the most vital process in the experiment. Examples of applying the femtosecond pump-probe technique is presented to study the relaxation dynamics of the molecular excited states, by what, on the basis of experimental conditions and requirements, the paper introduced approaches of determine the cross correlation function as well as the zero of time of the pump and probe laser pulse in the course of concrete femtosecond time-resolved experiments. For the pump and the probe laser pulses in the visible light range, by way of delay time of the pump and the probe pulses, we could apply optical technique to detect the transient of the intensity of the second harmonic generation. Yet, for the determination of correlation function as well as the zero of time for the ultrafast pulses in the UV or the VUV, calibration method would be an ideal method.
femtosecond laser; pump-probe; cross-correlation function; time-resolved
2016-11-07;
2017-01-19
国家自然科学基金(11564040,11204264); 新疆自治区青年科技创新人才培养工程(2013731012); 新疆师范大学重点实验室招标课题(KWFG1502)资助项目 Sopported by National Natural Science Foundation of China(11564040,11204264); Science and Technology Innovation Youth Talents Training Project of Xinjiang Autonomous Region(2013731012); Bidding Project for Key Laboratory of Xinjiang Normal University(KWFG1502)
1000-7032(2017)05-0648-07
O64
A
10.3788/fgxb20173805.0648
*CorrespondingAuthor,E-mail:maryam917@163.com
猜你喜欢
人人健康(2021年16期)2021-12-01
汕头大学学报(自然科学版)(2020年4期)2020-12-14
制造技术与机床(2019年8期)2019-09-03
华东师范大学学报(自然科学版)(2019年3期)2019-06-24
电子制作(2018年9期)2018-08-04
长春理工大学学报(自然科学版)(2018年2期)2018-05-26
中国设备工程(2017年24期)2017-12-28
中国光学(2016年2期)2016-11-09
系统工程与电子技术(2016年2期)2016-04-16
原子与分子物理学报(2015年3期)2015-11-24
