
多实验平台下基因及异构体表达分析综述
2017-06-01 12:20:40王凯莉刘学军
中国生物医学工程学报 2017年2期
王凯莉 张 礼 刘学军*
1(南京航空航天大学计算机科学与技术学院,南京 211106)2(南京林业大学信息科学技术学院,南京 210037)
多实验平台下基因及异构体表达分析综述
王凯莉1张 礼2刘学军1*
1(南京航空航天大学计算机科学与技术学院,南京 211106)2(南京林业大学信息科学技术学院,南京 210037)
转录组学研究近几年成为生命科学和医学领域的研究热点,基因表达水平测量则是转录组学研究的基础。差异基因表达分析对于了解基因功能具有重要作用,而差异异构体表达分析则能够反映选择性剪切变化的情况。当前大规模测量基因表达水平的实验平台主要包括基因芯片,以及基于高通量测序技术的RNA-Seq。首先介绍广泛使用的Affymetrix传统3′基因芯片、外显子芯片、较新的全转录组芯片,以及基于RNA-Seq技术的Illumina平台4个主流实验平台的技术原理;其次从基因表达水平计算和差异表达分析两方面介绍每个平台下一些主流数据分析方法和该研究设计的方法,分析每个平台下各数据分析方法的优劣,并进一步展示在标准数据集上一些代表性方法的对比结果。
传统3′基因芯片; 外显子芯片; HTA2.0芯片; RNA-Seq; 基因表达分析
引言
近年来,随着大规模基因表达水平测量技术的发展,逐渐形成两种主要测量技术,分别是基于杂交原理的基因芯片技术[1]和基于高通量测序技术的RNA-Seq[2]。其中,采用基因芯片技术的Affymetrix公司制备的传统3′基因芯片、外显子芯片、较新的全转录组芯片,以及采用RNA-Seq技术的Illumina测序平台均是被广泛使用的大规模基因表达水平测量平台。在基因及异构体表达分析中,差异表达(differential expression, DE)分析是最基本的研究目标之一。基因芯片早期在差异表达分析中占据绝对领先地位,但随着RNA-Seq技术蓬勃发展,RNA-Seq也被广泛应用于基因表达水平测量和差异表达分析。相比基因芯片,RNA-Seq并不依赖现有的基因注释信息而能获得几乎所有表达的转录,而基因芯片需要已知基因注释信息来设计探针,因此基因芯片无法检测到新的剪切异构体。此外,RNA-Seq背景噪声低,提高了表达水平测量的灵敏度和特异性。目前国际上对基因芯片与RNA-Seq的性能对比已有了一定的研究,如文献[3-4]对传统3′基因芯片与RNA-Seq在基因表达水平测量方面进行了对比研究。文献[5]主要在基因表达水平测量方面,对传统3′基因芯片、外显子芯片与RNA-Seq进行对比研究。文献[6]主要在基因表达水平测量和差异表达分析方面,对传统3′基因芯片和RNA-Seq进行对比研究。但现有的研究中较少考虑较新的全转录组芯片,并且在异构体差异表达分析方面的平台对比研究较少。
本研究全面综述了目前较常用的基因和异构体表达水平测量平台及其数据分析方法。首先介绍了传统3′基因芯片、外显子芯片、全转录组芯片以及基于RNA-Seq技术的Illumina平台的技术原理。其次从基因表达水平计算和差异表达分析两方面介绍了每个平台下一些主流数据分析方法和笔者设计的方法,分析了每个平台下各数据分析方法的优劣,并进一步展示了在生物芯片质量控制项目(microarray quality control, MAQC)的系列标准数据集[7-9]上一些代表性方法的对比结果,为生物医学领域研究学者对于实验平台和数据分析方法的选择提供参考。
1 平台介绍
1.1 基因芯片
基因芯片是20世纪90年代建立起来的大规模基因表达水平测量技术,该技术基于杂交原理,主要特点是高通量、自动化和微型化。Affymetrix公司的芯片产品非常丰富,并被业界广泛采用。目前Affymetrix公司的基因芯片在公共数据库(如GEO)中累积了大量的数据,以满足学者们深入研究的需要[10]。基因芯片实验步骤如下:首先用荧光标记物标记待测样本,并放入基因芯片自动孵育装置中进行杂交;杂交完成后,检测探针的信号强度,即探针检测到样本的表达量;最后将实验结果保存到细胞密度文件(CEL文件)中,即基因芯片原始数据。
Affymetrix公司的传统3′基因芯片包含130万个探针,具有独特的PM-MM探针对。PM(perfect-match)探针的碱基序列和目标序列完全匹配,MM(mis-match)探针仅将序列中间的一位碱基换成互补碱基。芯片上每个基因对应一个或多个探针集,这些探针集由25碱基长度的PM-MM探针对构成,提高了对低表达样本的检测[11]。本研究采用了人类基因芯片Human Genome U133 Plus 2.0 Array(U133)。
Affymetrix公司的外显子芯片仅设计了PM探针[12]。人类外显子芯片包含550万个探针,构成大约140万个探针集,每个基因平均覆盖40个探针,每个外显子平均覆盖4个探针(见图1)。传统3′基因芯片的探针仅覆盖转录组的3′区域,而外显子芯片的探针覆盖了所有编码转录组,覆盖范围更广,准确性更高。本研究实验中采用的外显子芯片是Human Exon 1.0 ST Array(Exon array)。
Affymetrix公司在2013年推出一款人类全转录组基因芯片(Human Transcriptome Array 2.0, HTA2.0)[13]。该芯片包含外显子探针、外显子之间的剪切结合区探针、SNP探针、lncRNA探针等多种探针,总数近700万个。每个外显子覆盖约10个探针,外显子之间的剪切结合区覆盖4个探针(见图1),可检测超过24万条编码转录本和超过4万条非编码转录本。
传统3′基因芯片的探针设计注重转录组的3′区域(见图1),探针覆盖范围较窄,而且也未考虑基因的选择性剪切,因此无法测量异构体表达水平,应用范围受到一定的限制。而外显子芯片的探针覆盖范围较广,可用于基因、外显子和剪切异构体水平分析。HTA2.0芯片探针类型多样,覆盖范围更为全面,不仅应用于基因、外显子和剪切异构体水平分析,还应用于非编码转录组分析等,具有最为广泛的应用前景。
1.2 RNA-Seq技术
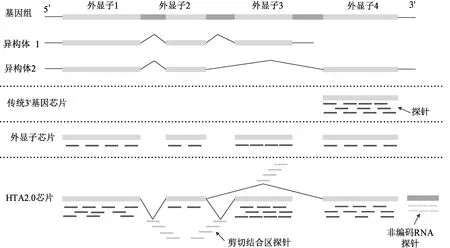
图1 传统3′基因芯片、外显子芯片和HTA2.0芯片的探针设计对比Fig.1 Comparison of probe design of traditional 3′ GeneChip, Exon array and HTA2.0
RNA-Seq是基于高通量测序技术对转录组进行研究的新一代测序方法,并以高通量,所需样本少等优势,迅速成为研究基因及异构体表达水平的主流方法[3]。RNA-Seq技术可应用于全基因组范围内基因的表达水平测量和差异表达分析,并具有定量研究选择性剪切[14-15],发现未知序列特征的能力。
RNA-Seq实验一般过程如图2所示,主要分为以下几个步骤:首先选择mRNA,并片段化。其次将mRNA片段逆转录成cDNA,并进行PCR 扩增。之后将cDNA片段的两端加上接头得到测序需要的文库。最后采用测序平台进行测序。经过激光照射和图像分析,获得被测碱基和质量评分,并将同一位置的碱基根据测序顺序连成读段(read),这就是RNA-Seq测序后得到的最原始数据[16]。目前,Roche公司的454技术、Illumina公司的Solexa技术以及ABI公司的SOLiD技术等测序技术被广泛使用,其中使用最为广泛的是Illumina/Solexa测序平台。
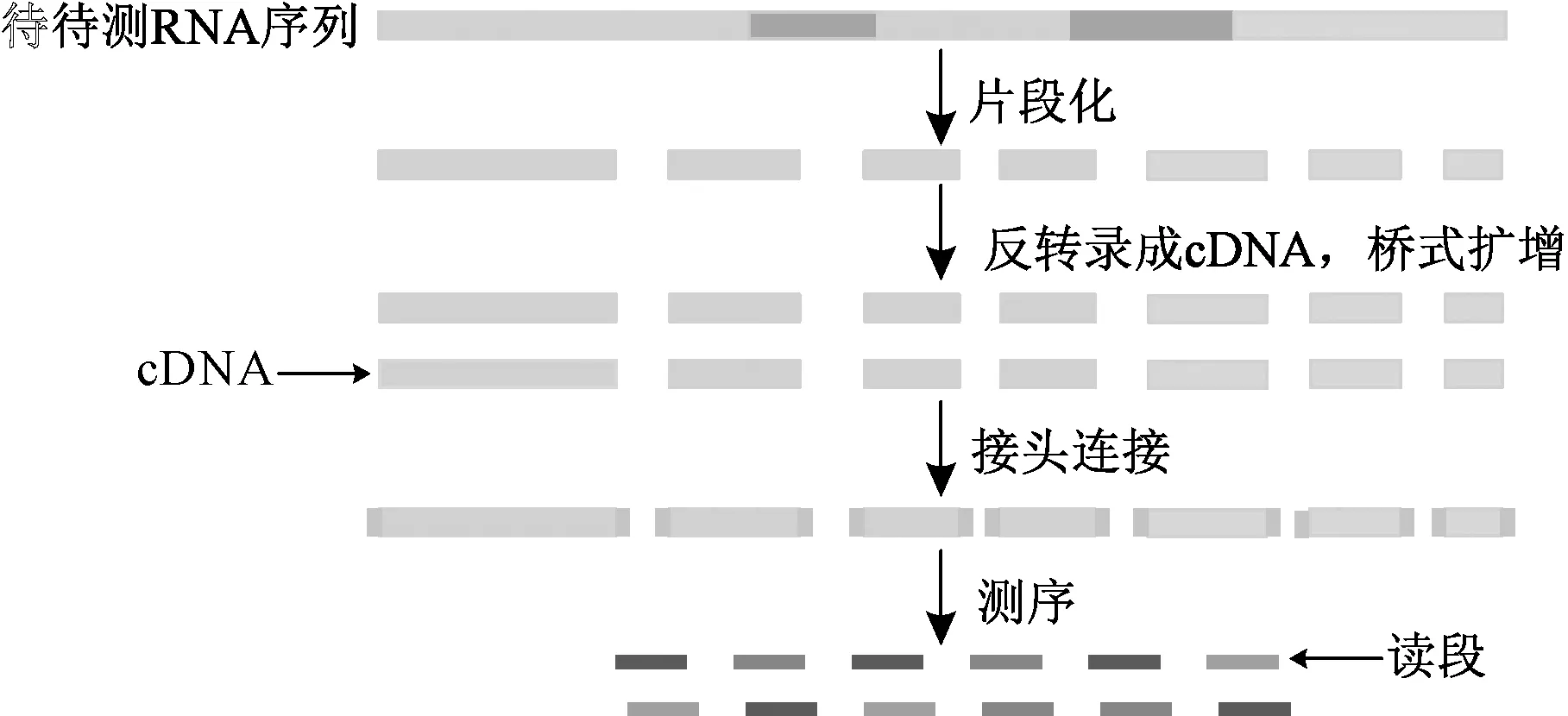
图2 RNA-Seq 实验一般过程Fig.2 Process of RNA-Seq experiment
2 基因表达水平计算
2.1 基因芯片原始数据分析
基因芯片数据分析过程如图3所示。首先从基因芯片原始数据中提取出探针的灰度值,然后根据基因或剪切异构体以及探针的注释文件,利用数据分析方法,计算基因或异构体的表达水平,为后续分析提供准确的数据。基因芯片数据分析的主要困难是探针的非特异杂交特性,导致原始数据中存在大量的噪声。另外,基因芯片数据分析方法依赖于基因注释信息,其完善程度会对分析结果产生很大的影响。目前,许多学者提出了各种算法对原始数据进行去噪处理,如传统算法RMA[17],以及本研究小组已发表的伽马模型mmgMOS[18]和GME[19]等,下载网址如表1所示。

图3 基因芯片数据分析过程Fig.3 Process of microarray data analysis
表1 基因芯片及RNA-Seq的计算基因或异构体表达水平的方法列表
Table 1 Methods and softwares to calculate gene and isoform expression levels for microarray and RNA-Seq

平台计算方法下载网址基因芯片RMAhttp://www.bioconductor.org/packages/oligo.htmlmmgMOShttp://www.bioconductor.org/packages/puma.htmlGMEhttp://www.bioconductor.org/packages/puma.htmlRNA-SeqCufflinkshttp://cole-trapnell-lab.github.io/cufflinks/MMSEQhttps://github.com/eturro/mmseqkallistohttps://github.com/pachterlab/kallistoStringTiehttps://github.com/gpertea/stringtiePGSeqhttps://github.com/PUGEA/PGSeq
RMA算法仅采用PM探针的灰度值来计算基因表达水平。为了消除噪声的影响和保持数据的一致性,进行背景校正和归一化。之后利用经过背景校正及归一化后探针的灰度值拟合一个线性相加模型以获得基因的表达水平。由于选择性剪切,一个基因会对应一个或多个剪切异构体,而在基因芯片上一个剪切异构体往往对应多个探针,有些探针可以被不同的剪切异构体所共享(见图1)。这种基因、剪切异构体及探针的多元映射关系,导致了获取异构体对应的探针灰度值时具有很高的不确定性,给异构体表达水平的计算带来挑战。由于RMA算法无法处理基因、剪切异构体及探针的多元映射,因此只能计算基因的表达水平,无法计算异构体的表达水平。目前,RMA 算法实现在生物信息学组件Bioconductor中的oligo软件包中,如表1所示。mmgMOS是基于伽马分布并针对多重复芯片的概率模型,用于传统3′基因芯片数据分析。与RMA算法不同,mmgMOS概率模型能够很好地模拟基因芯片实验中的不确定性,抗噪能力强,同时采用PM和MM探针的灰度值,并考虑了两者之间的相关性,提高了基因表达水平计算的准确性。GME模型也是基于伽马分布的概率模型,根据基因、剪切异构体及探针的多元映射关系,计算基因和异构体表达水平,并特别地考虑了异构体所共享的探针。与RMA等传统的计算方法相比,GME的优势在于能够同时计算基因和异构体的表达水平,因此可以更好地应用于选择性剪切的研究。另外,GME模型可以获得表达水平的不确定度,提高了差异表达分析的准确性。根据GATExplorer[20]和Microarray Lab[21]分别提供的外显子芯片和HTA2.0芯片的注释文件,提取出基因、剪切异构体及探针的映射关系,因此GME模型适用于外显子芯片和HTA2.0芯片。目前,mmgMOS和GME实现在Bioconductor的puma软件包中。
2.2 RNA-Seq原始数据分析
RNA-Seq原始数据分析过程如图4所示。首先采用序列对比方法将读段定位到参考基因组或转录组上;然后通过计数映射到基因及其异构体上的读段数目来计算基因及异构体表达水平。表达水平计算的主要困难是读段的多源映射问题和读段在参考序列上呈非均匀分布。读段的多源映射问题一方面是由于RNA-Seq实验产生的读段通常较短,一般是25~400 bp(base pairs),而转录组长度很长,一般包含了上万个碱基,这样读段无法完全覆盖转录组,从而导致相当一部分读段在参考基因组上有多个匹配位点[22]。随着测序技术的发展,读段长度不断增加或者制备双末端读段等方式可以降低读段多源映射的影响。另一方面由于真核生物普遍存在选择性剪切现象,同一个读段会被基因的不同剪切异构体所共享,从而不能将读段准确地映射到单一异构体上[23],为异构体表达水平的计算增加了难度。
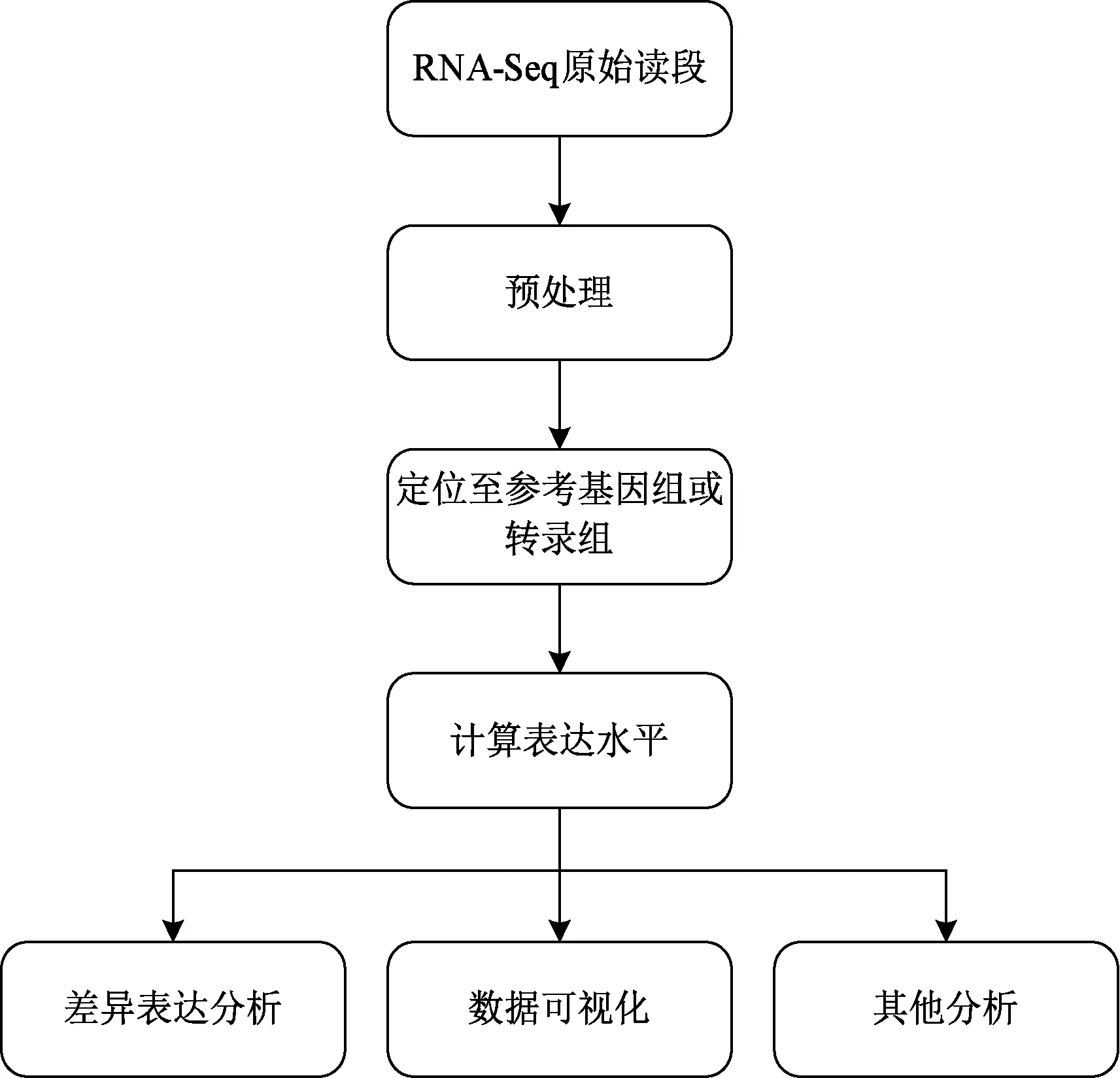
图4 RNA-Seq数据分析过程Fig.4 Process of RNA-Seq data analysis
读段的非均匀分布是由于在制备cDNA文库时人为地引入了一些偏好,如在RNA逆转录cDNA的过程中加入的随机引物对不同的RNA会产生不同程度的偏好[16]。另外,对RNA-Seq原始数据处理不当也会导致读段在参考序列上非均匀分布,如直接丢弃多源映射读段和低质量分数的读段等。当前有学者提出了很多方法来解决这些问题,如主流方法Cufflinks[24],较新的MMSEQ[25]、kallisto[26]和StringTie[27],以及本研究小组已提出的方法PGSeq[28]等,上述方法下载网址如表1所示。
Cufflinks方法采用泊松分布模拟读段在外显子上的分布,消除读段多源映射的影响,同时对读段非均匀分布偏好的随机特性进行模拟。MMSEQ方法采用泊松-伽马双层模型来模拟异构体随机表达的特性,消除读段多源映射的影响。kallisto方法采用读段到转录组的伪比对策略,能够快速地定量分析异构体表达水平,从而获得基因表达水平。StringTie采用网络流算法和可选的从头(de novo)组装转录组并估算表达水平,能够在拼接出转录组的同时进行表达水平的定量分析。PGSeq方法采用泊松分布来模拟映射到每个外显子上的读段数,消除读段多源映射的影响,同时引入了伽玛因子来模拟读段非均匀分布的偏好信息。虽然MMSEQ与 PGSeq方法均采用泊松-伽马分布双层模型,但是MMSEQ并未考虑读段在参考序列上呈非均匀分布,而PGSeq方法引入伽马分布的隐含变量来模拟读段的非均匀分布特性,并推导出基因及异构体的表达水平服从负二项分布,能够更好地模拟读段数据的散布特点,提高了计算准确性。表2显示了在MAQC数据集下4个平台的不同表达水平计算方法的准确性。MAQC数据集提供的804个qRT- PCR验证基因作为基准,在通用人类参考RNA(universal human reference RNA, UHRR)和人类大脑参考 RNA(human brain reference RNA, HBRR)两个条件下进行比较。不同方法计算得到的基因表达值与qRT-PCR实验获得的基因表达值的相关系数(squared Pearson correlation coefficient, R2)被用来评价准确性,相关系数越接近1,则说明测量结果的准确度越高。为避免较大的表达水平对相关系数的影响,本研究对所有基因的表达水平进行对数转换后再计算相关系数。
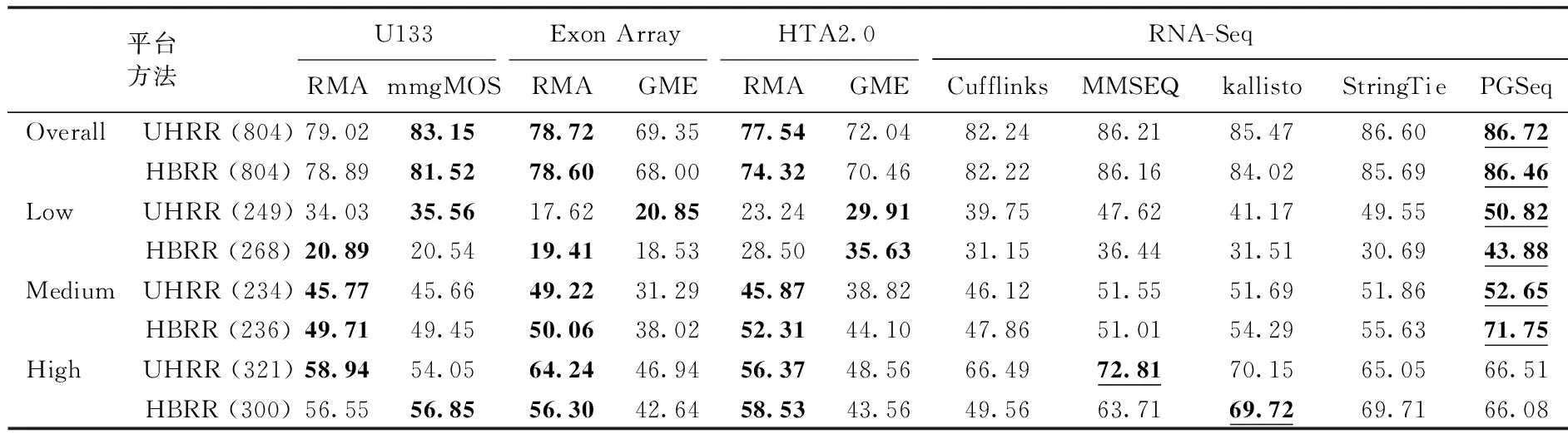
表2 在MAQC数据集的UHRR和HBRR 2个条件下4个平台的不同表达水平计算方法的准确性(%)Tab.2 Accuracy (%) of various gene expression calculation methods for UHRR and HBRR conditions under MAQC
注:根据qRT-PCR测量值,804个qRT-PCR验证的基因被划分为3个区间,分别是低表达区间(Low),中表达区间(Medium)和高表达区间(High)。括号里的数字代表在两个样本下相应表达区间的qRT-PCR验证的基因数目。
Note: According to qRT-PCR measurements 804 genes are divided into three intervals, Low, Medium and High. The numbers in brackets represent the numbers of genes that are validated by qRT-PCR in UHRR and HBRR samples.
3 差异表达分析
在基因芯片和RNA-Seq数据分析中,差异表达分析是最基本的研究目标,通过分析不同条件下的转录组表达数据,识别发生差异表达的基因或异构体,这对揭示基因调控规律或基因选择性剪切的变化具有重要作用。
3.1 基因芯片差异表达分析
由于基因芯片发展时间较长,人们已经提出很多差异表达分析方法。例如,limma[29]、PBR[30]以及研究小组已发表的PPLR方法[31],其下载网址如表3所示。limma方法应用范围较为广泛,其核心思想就是用一个线性模型来拟合每个基因的表达数据。limma方法适用于基因芯片和RNA-Seq等平台。PBR(Penalized Binomial Regression)是基于PED(Penalized Euclidean Distance)的惩罚二项式回归算法。首先,利用PED对基因表达数据进行分类并排序;其次,利用真实已知的数据进行仿真,识别差异表达的基因和异构体。PBR方法与limma相同,适用于基因芯片和RNA-Seq等平台。但是,这些方法忽略了很多潜在且有用的信息,如表达水平的技术性测量误差。若能够从原始数据获得更多的先验信息,可以提高模型的性能。因此,PPLR方法采用贝叶斯模型,并考虑了表达水平的不确定度,从而提高了差异检测的准确度。
表3 基因芯片及RNA-Seq的差异表达分析方法
Table 3 Methods and softwares to detect DE genes and isoforms for microarray and RNA-Seq
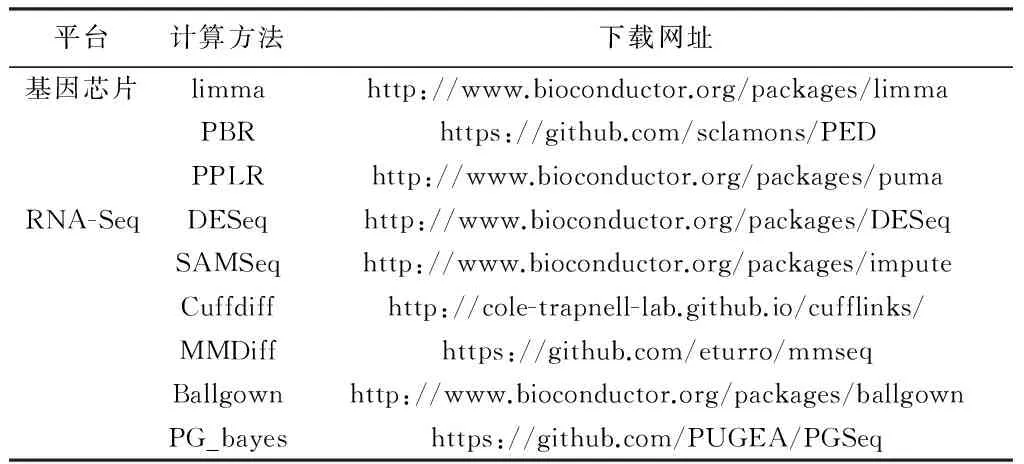
平台计算方法下载网址基因芯片limmahttp://www.bioconductor.org/packages/limmaPBRhttps://github.com/sclamons/PEDPPLRhttp://www.bioconductor.org/packages/pumaRNA-SeqDESeqhttp://www.bioconductor.org/packages/DESeqSAMSeqhttp://www.bioconductor.org/packages/imputeCuffdiffhttp://cole-trapnell-lab.github.io/cufflinks/MMDiffhttps://github.com/eturro/mmseqBallgownhttp://www.bioconductor.org/packages/ballgownPG_bayeshttps://github.com/PUGEA/PGSeq
3.2 RNA-Seq差异表达分析
RNA-Seq差异表达分析方法主要分为两种,一是基于读段计数方法,二是两步法。在读段计数方法中,有DESeq[32]和SAMSeq等[33]。DESeq采用负二项分布,解决了读段非均匀的问题;非参数模型SAMSeq仅对基因表达水平排序,以识别差异表达的基因。这类方法可以有效地识别差异表达的基因,但是不能直接用来识别差异表达的异构体。两步法能够同时识别差异表达的基因和异构体,使用范围更加广泛,如Cufflinks和Cuffdiff[34]、MMSEQ和MMDiff[35]、kallisto和limma、StringTie和Ballgown[36]以及PGSeq和PG_bayes[37]等,上述方法下载网址如表3所示。Cuffdiff使用 Cufflinks 方法得到表达水平,并使用一个线性模型识别差异表达的基因和异构体。MMDiff方法克服了数据高度结构化的问题,并考虑了表达水平的不确定度。Ballgown方法是基于F-test识别差异表达的基因和异构体。与Cuffdiff相似,Ballgown可以处理Cufflinks输出的表达数据,但是其效率和准确度高于Cuffdiff。另外,本研究小组已发表的差异检测方法PG_bayes根据模型选择的思想,基于PGSeq方法推导出的表达水平的负二项分布模型,采用贝叶斯因子方法,并考虑了表达水平的不确定度,从而提高差异表达分析的灵敏度和准确度。图5和表4显示了3种基因芯片和RNA-Seq在MAQC数据集上获得的121个基因的差异表达分析结果。由于传统3′基因芯片无法测量异构体表达水平,因此图6和表5显示了外显子芯片、HTA2.0芯片和RNA-Seq在MAQC数据集上获得的529个异构体的差异表达分析结果(见下页)。

表4 不同平台下121个共同基因差异表达分析的AUCTable 4 AUC of DE gene analysis for the various platforms
注:RMA、mmgMOS和GME使用的差异表达分析方法均是PPLR。Cufflinks、MMSEQ、kallisto、StringTie和PGSeq使用的差异表达分析方法分别是Cuffdiff、MMDiff、limma、Ballgown和PG_bayes。
Note: RMA, mmgMOS and GME use PPLR for DE analysis. Cufflinks, MMSEQ, kallisto, StringTie and PGSeq use Cuffdiff, MMDiff, limma, Ballgown and PG_bayes for DE analysis, respectively.
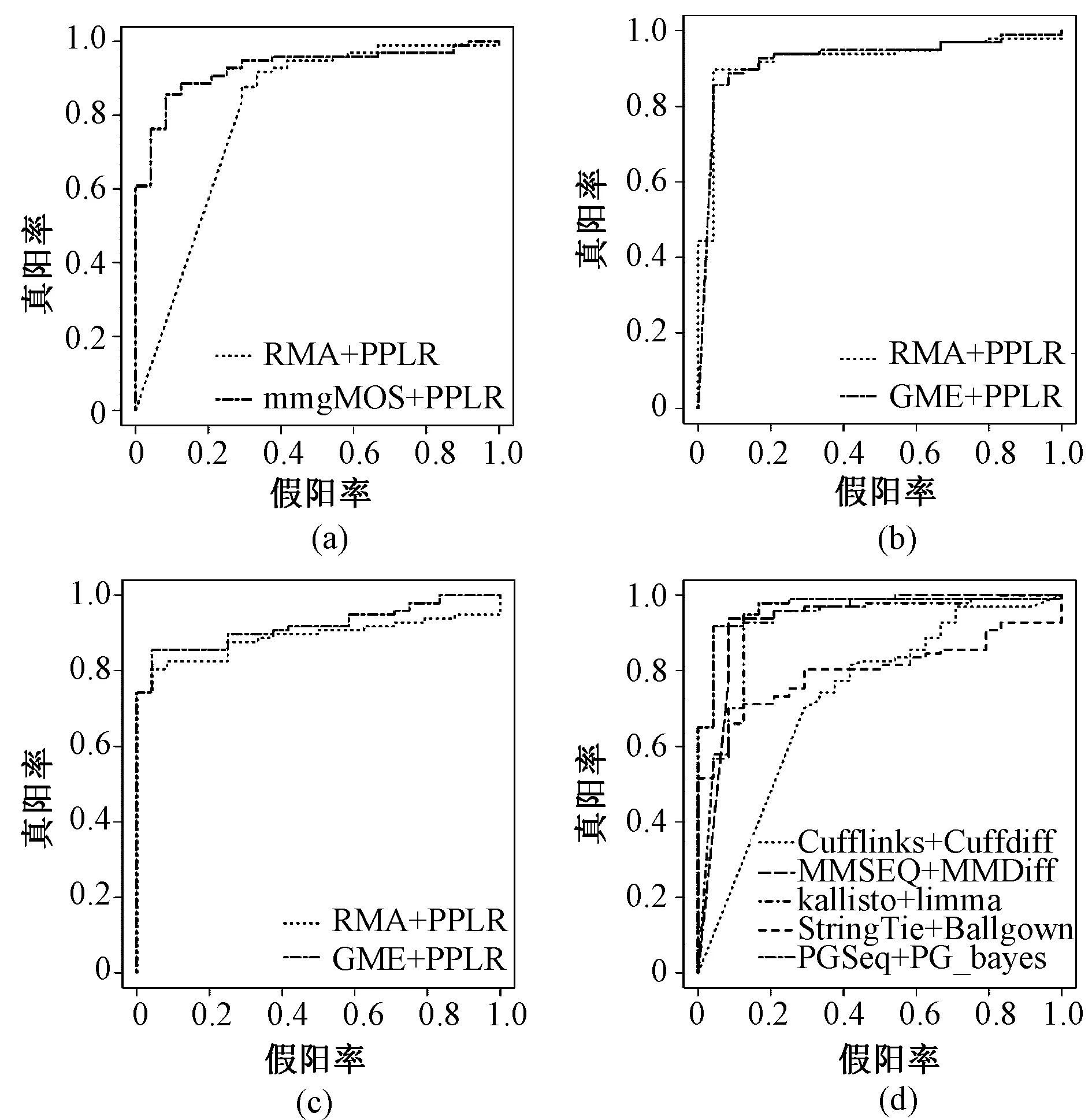
图5 4个平台下121个共同基因差异表达分析的ROC曲线。(a)U133;(b)外显子芯片;(c)HTA2.0;(d)RNA-SeqFig.5 ROC curves of DE analysis for the 121 common genes found on the four platforms. (a) U133; (b) Exon array; (c) HTA2.0; (d) RNA-Seq
4 总结与讨论
本研究对传统3′基因芯片、外显子芯片、HTA2.0芯片及RNA-Seq技术进行了总结,并从基因表达水平测量和差异表达分析两方面,介绍了这4种平台下具有代表性或较新的数据分析方法,并进一步展示了一些方法在MAQC数据集上的对比结果,为不同需求的研究学者对于实验平台和数据分析方法的选择提供参考。虽然RNA-Seq技术在转录组数据分析中具有一定的优势,但是RNA-Seq分析结果的准确性依赖于测序深度,测序深度增加,成本也会增加,而基因芯片由于实验成本相对较低,在大规模已知序列的基因表达分析中,基因芯片仍具有很大的应用空间。研究者可以根据不同的研究目的以及研究成本等因素,选择合适的实验平台。此外,不同平台下不同的数据分析方法对分析结果的准确性也有重要影响。
由于测序技术的快速发展,大量可用的方法用于基因芯片和RNA-seq数据分析。选择合适的数据分析方法进行不同的研究并选择最佳参数都是至关重要的,这些因素直接影响最终结果和生物过程的解释。另外,对于基因芯片和RNA-seq数据分析,注释文件或参考基因组序列对于分析过程和结果具有重要影响。由于基因芯片数据分析方法依赖于基因、异构体以及探针的注释信息,其完善程度会对分析结果影响较大。例如,传统3′基因芯片、外显子芯片和HTA2.0芯片,均采用RMA算法计算基因表达水平,但是HTA2.0芯片的准确率略低于其他两种芯片的准确率。由于HTA2.0芯片是一种相对较新的芯片,其注释信息不够完善是导致其准确率低于传统3′基因芯片和外显子芯片的主要原因。在RNA-Seq数据分析过程中,参考基因组注释的完备程度也会影响后续分析的策略和准确性。当参考基因组注释比较完备时(如人类和老鼠),读段可以直接定位到参考基因组序列,在后续分析中不需要进行转录组重构。但是,参考基因组序列注释不完善,后续分析中需要进行转录组重构,以提高分析结果的准确性。另外,由于绝大部分生物是没有参考基因组序列或者参考基因组序列的可信度较低,在后续分析过程就需要采取从头组装转录组,提高后续分析的准确性。因此,根据不同的研究目标,除了选择合适的实验平台外,选取准确的数据分析方法和注释文件或参考基因组序列,对获得合理的分析结果同样重要。

表5 3个平台异构体差异表达分析的AUCTable 5 AUC of DE isoform analysis for the three platforms
注: GME使用的差异表达分析方法是PPLR,Cufflinks、MMSEQ、kallisto、StringTie和PGSeq使用的差异表达分析方法分别是Cuffdiff、MMDiff、limma、Ballgown和PG_bayes。
Note: GME uses PPLR for DE analysis. Cufflinks, MMSEQ, kallisto, StringTie and PGSeq use Cuffdiff, MMDiff, limma, Ballgown and PG_bayes for DE analysis, respectively.
[1] Schena M, Shalon D, Davis RW, et al. Quantitative monitoring of gene expression patterns with a complementary DNA microarray [J]. Science, 1995, 270(5235): 467-470.
[2] Wang Zhong, Gerstein M, Snyder M. RNA-Seq: a revolutionary tool for transcriptomics [J]. Nature Reviews Genetics, 2009, 10(1): 57-63.
[3] Marioni JC, Mason CE, Mane SM, et al. RNA-seq: an assessment of technical reproducibility and comparison with gene expression arrays [J]. Genome Research, 2008, 18(9): 1509-1517.
[4] Xu Xiao, Zhang Yuanhao, Williams J, et al. Parallel comparison of Illumina RNA-Seq and Affymetrix microarray platforms on transcriptomic profiles generated from 5-aza-deoxy-cytidine treated HT-29 colon cancer cells and simulated datasets. BMC Bioinforma 14:S1 [J]. Bmc Bioinformatics, 2013, 14(9):1-14.
[5] Bemmo A, Benovoy D, Kwan T, et al. Gene expression and isoform variation analysis using Affymetrix exon arrays [J]. Bmc Genomics, 2008, 9(1):1-15.
[6] Zhao Shanrong, Fung-Leung Wai-Ping, Bittner A, et al. Comparison of RNA-Seq and microarray in transcriptome profiling of activated T cells [J]. PLoS ONE, 2014, 9(1): e78644.
[7] Shi Leming, Reid LH, Jones WD, et al. The MicroArray Quality Control (MAQC) project shows inter-and intraplatform reprodu-cibility of gene expression measurements [J]. Nature Biotechnology, 2006, 24(9): 1151-1161.
[8] MAQC Consortium. The MicroArray Quality Control (MAQC)-II study of common practices for the development and validation of microarray-based predictive models [J]. Nature Biotechnology, 2010, 28(8): 827-838.
[9] Seqc/Maqc-Iii Consortium. A comprehensive assessment of RNA-seq accuracy, reproducibility and information content by the Sequencing Quality Control Consortium [J]. Nature Biotechnology, 2014, 32(9): 903-914.
[10] Dalma-Weiszhausz DD, Warrington J, Tanimoto EY, et al. The Affymetrix GeneChip© Platform: An Overview [J]. Methods in Enzymology, 2006, 410: 3-28.
[11] Southern E, Mir K, Shchepinov M. Molecular interactions on microarrays [J]. Nature Genetics, 1999, 21(1 Suppl):5-9.
[12] Affymetrix: Affymetrix Gene Chip exon array design [R]. 2005.
[13] Affymetrix: GeneChip Human Transcriptome Array 2.0 [R]. 2013.
[14] Valenzuela A, Talavera D, Orozco M, et al. Alternative splicing mechanisms for the modulation of protein function: conservation between human and other species [J]. Journal of Molecular Biology, 2004, 335(2): 495-502.
[15] Wang ET, Sandberg R, Luo S, et al. Alternative isoform regulation in human tissue transcriptomes [J]. Nature, 2008, 456(7221): 470-476.
[16] 王曦, 汪小我, 王立坤, 等. 新一代高通量 RNA 测序数据的处理与分析[J]. 生物化学与生物物理进展, 2010, 37(8): 834-846.
[17] Irizarry RA, Hobbs B, Collin F, et al. Exploration, normalization, and summaries of high density oligonucleotide array probe level data [J]. Biostatistics, 2003, 4(2): 249-264.
[18] Liu Xuejun, Milo M, Lawrence ND, et al. A tractable probabilistic model for Affymetrix probe-level analysis across multiple chips [J]. Bioinformatics, 2005, 21(18): 3637-3644.
[19] Liu Xuejun, Gao Zhenzhu, Zhang Li, et al. puma 3.0: improved uncertainty propagation methods for gene and transcript expression analysis [J]. Bmc Bioinformatics, 2013, 14(3):1-15.
[21] Dai Manhong, Wang Pinglang, Boyd AD, et al. Evolving gene/transcript definitions significantly alter the interpretation of GeneChip data [J]. Nucleic Acids Research, 2005, 33(20): e175-e175.
[22] Pasaniuc B, Zaitlen N, Halperin E. Accurate estimation of expression levels of homologous genes in RNA-seq experiments [J]. Journal of Computational Biology, 2011, 18(3): 459-468.
[23] Costa V, Angelini C, De FI, et al. Uncovering the Complexity of Transcriptomes with RNA-Seq [J]. Biomed Research International, 2010, 2010(1):853916.
[24] Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA-Seq reveals unannotated transcripts and isoform switching during cell differentiation [J]. Nature Biotechnology, 2010, 28(5): 511-515.
[25] Turro E, Su SY, Gonçalves, et al. Haplotype and isoform specific expression estimation using multi-mapping RNA-seq reads [J]. Genome Biology, 2011, 12(2):81-89.
[26] Bray NL, Pimentel H, Melsted P, et al. Near-optimal probabilistic RNA-seq quantification [J]. Nature Biotechnology, 2016, 34(5):525 -527.
[27] Pertea M, Pertea GM, Antonescu CM, et al. StringTie enables improved reconstruction of a transcriptome from RNA-seq reads [J]. Nature Biotechnology, 2015, 33(3): 290-295.
[28] Liu Xuejun, Zhang Li, Chen Songcan. Modeling exon-specific bias distribution improves the analysis of RNA-seq data [J]. PLoS ONE, 2015, 10(10): e0140032.
[29] Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA-sequencing and microarray studies [J]. Nucleic Acids Research, 2015, 43(7):e47.
[30] Vasiliu D, Clamons S, Mcdonough M, et al. A regression-based differential expression detection algorithm for microarray studies with ultra-low sample size [J]. PLoS ONE, 2015; 10(3): e0118198.
[31] Liu Xuejun, Milo M, Lawrence ND, et al. Probe-level measurement error improves accuracy in detecting differential gene expression [J]. Bioinformatics, 2006, 22(17): 2107-2113.
[32] Anders S, Huber W. Differential expression analysis for sequence count data [J]. Genome Biology, 2010, 11(10):1-12.
[33] Li Jun, Tibshirani R. Finding consistent patterns: A nonparametric approach for identifying differential expression in RNA-Seq data [J]. Statistical Methods in Medical Research, 2013, 22(5):519-536.
[34] Trapnell C, Roberts A, Goff L, et al. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks [J]. Nature Protocols, 2012, 7(3): 562-578.
[35] Schweikert G, Cseke B, Clouaire T, et al. MMDiff: quantitative testing for shape changes in ChIP-Seq data sets [J]. Bmc Genomics, 2013, 14(48):5492-5500.
[36] Frazee AC, Pertea G, Jaffe AE, et al. Ballgown bridges the gap between transcriptome assembly and expression analysis [J]. Nature Biotechnology, 2015, 33(3): 243-246.
[37] 王黎, 刘学军, 张礼. 基于模型选择的差异基因和异构体检测[J]. 数据采集与处理,2016,31(5):965-973.
A Review of Gene and Isoform Expression Analysis across Multiple Experimental Platforms
Wang Kaili1Zhang Li2Liu Xuejun1*
1(CollegeofComputerScienceandTechnology,NanjingUniversityofAeronauticsandAstronautics,Nanjing211106,China)2(CollegeofInformaitonScienceandTechnology,NanjingForestryUniversity,Nanjing210037,China)
Transcriptomics study has become a hot topic in life science and medical research in recent years. From the expression point of view, the foundation of transcriptomics study is the measurement of gene expression levels. Differential expression (DE) analysis of genes is very important for understanding the function of genes. DE analysis of isoforms is a feasible method to reflect the change of alternative splicing. Currently, there are mainly two large-scale experimental platforms for measuring gene expression levels, including microarray and high-throughput sequencing technology, RNA-Seq. At the beginning of this paper, we introduced the technical principles of the four mainstream experimental platforms: Affymetrix′s traditional 3′ GeneChip, Exon array, Human Transcriptome Array 2.0 and Illumina platform based on RNA-Seq. We then reviewed the mainstream analysis methods and our methods on each platform for the calculation of gene expression levels and DE analysis. We also showed the comparison results of expression measurement and DE analysis across various platforms under a well-defined benchmark data set.
traditional 3′ GeneChip; Exon array; HTA2.0; RNA-Seq; gene expression analysis
10.3969/j.issn.0258-8021. 2017. 02.012
2016-04-17, 录用日期:2016-10-23
国家自然科学基金(61170152)
R318
A
0258-8021(2017) 02-0211-08
*通信作者(Corresponding author),E-mail: xuejun.liu@nuaa.edu.cn
猜你喜欢
系统仿真技术(2022年4期)2023-01-17 13:01:44
电子科技大学学报(2022年5期)2022-10-29 01:57:52
云南化工(2021年8期)2021-12-21 06:37:38
今日农业(2021年4期)2021-06-09 06:59:56
中国生殖健康(2020年4期)2021-01-18 02:58:10
中国生殖健康(2018年4期)2018-11-06 07:12:16
现代检验医学杂志(2016年4期)2016-11-15 02:01:00
国外医药(抗生素分册)(2016年4期)2016-07-12 14:25:19
信息记录材料(2016年4期)2016-03-11 15:22:30
哈尔滨医药(2015年3期)2015-12-01 03:57:44
