
先天性肌营养不良1A型1例临床与基因分析
2017-05-24 14:45:57江士远
临床儿科杂志 2017年5期
江士远 向 娜
单县海吉亚医院儿科(山东单县 274300)
先天性肌营养不良1A型1例临床与基因分析
江士远 向 娜
单县海吉亚医院儿科(山东单县 274300)
目的报道1例LAMA2基因变异导致先天性肌营养不良的临床、实验室检查及遗传学特点。方法回顾分析1例先天性肌营养不良1A型患儿的临床资料,并复习相关文献。结果患儿,男,5岁2个月,临床表现为运动发育落后,2岁时可独坐,不能独走;肌力及肌张力低下,早期出现关节挛缩。生化检测发现肌酸激酶(CK)升高(491 U/L),其同工酶CK-MB升高(41.8 U/L);肌电图提示肌源性损害可能;头颅MRI提示大脑白质异常信号。基因检测发现LAMA2存在复杂杂合突变,c.2045-2046delAG杂合缺失,来自母亲,为已报道的致病变异;exon5存在杂合缺失,来自父亲,为未报道的新变异,软件功能预测提示为致病性变异。结论LAMA2基因变异导致先天性肌营养不良,患儿以运动发育落后起病,CK升高,高通量基因检测有助于明确诊断。
先天性肌营养不良; 临床特点; 分子诊断
先天性肌营养不良(congenital muscular dystrophy,CMD)是一类出生后早期起病、主要影响骨骼肌功能的疾病,不同的肌营养不良虽有相似的临床表型,但遗传差异很大[1]。LAMA2基因突变导致的先天性肌营养不良1A型(congenital muscular dystrophy type 1A, MDC1A)又称merosin缺乏症,是首先被分离出的一种肌营养不良症,约占先天性肌营养不良的40%,属于常染色体隐性遗传病,欧美国家较多见[2,3]。MDC1A临床表现为出生时或出生后不久出现肌张力低下、肌肉无力、运动发育迟缓以及关节挛缩等症状,肌酶可有轻到中度升高,肌肉病理检测可见肌营养不良改变[4]。现报告1例MDC1A,并对相关的文献进行复习,探讨临床表现以及基因检测对诊断的重要性。
1 临床资料
患儿,男,5岁2个月,足月剖宫产,母孕期及出生时未见异常,出生后喂养困难,2岁可独坐,早期出现关节萎缩,至今不能独走。就诊时体格检查:神清,眼距宽,鼻梁低,上下牙齿不合;双侧呼吸运动对称,未闻及啰音;心律齐,心音有力,心脏各听诊区均未闻及杂音;腹软,肝脾肋下未触及;四肢肌力及肌张力均降低,膝腱反射未引出,病理反射(-)。实验室检查:肌酸激酶(creatine kinase,CK)491 U/L(参考值25~195 U/L),肌酸激酶同工酶(creatinine kinase MB isoenzyme,CK-MB)41.8 U/L(参考值0~25 U/L);肌电图检测发现肌源性损害可能;头颅MRI提示大脑白质异常信号,主要累及侧脑室前后角。根据以上表现患儿拟诊为先天性肌营养不良1A型。其父母体健,非近亲结婚,双方家族中无类似疾病史。
经家属知情同意以及医院医学伦理委员会审核后,抽取患儿及其父母外周静脉血2 mL,置EDTA抗凝管,利用基因组DNA提取试剂盒(AXYGEN公司)提取基因组DNA,并测定浓度。委托华大基因公司利用目标区域序列捕获法对患儿进行高通量测序分析(先天肌营养不良相关的基因检测包),测序深度为200×,测序后行相应的软件分析。结果发现患儿LAMA2基因存在c.2045-2046delAG杂合缺失(图1),2个碱基缺失可导致基因所编码的蛋白产生移码突变即LAMA2蛋白在682位赖氨酸开始产生移码突变并在之后22位氨基酸处终止(p.Lys682fsX22)。

图1 突变位点高通量测序reads比对图
对突变结果进行Sanger测序验证。在LAMA2基因突变位点的两端设计扩增引物,利用PCR方法扩增出含有突变位点的片段,其中25μL的PCR扩增体系中含10 mmol Tris-HCl(pH 8.3),50 mmol KCl,1.5 mmol MgCl2,0.3 mmol dNTP,引物各200 pmol,Taq 1.0 U(东盛生物公司),在Bio-Rad PCR仪上进行扩增,扩增程序为:94℃ 5 min;94℃ 30 s,56℃ 30 s,72℃ 30 s,共34个循环。扩增产物测序发现患儿存在c.2045-2046delAG杂合缺失,其母亲也存在该位点突变,其父亲未见异常,见图2,说明患儿该位点的异常遗传自其母亲。
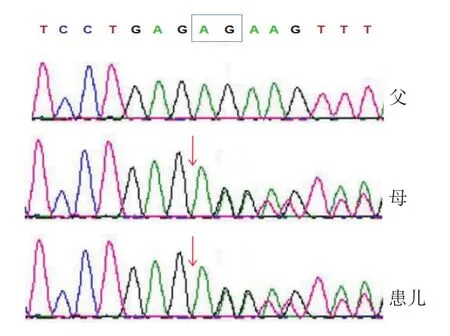
图2 c.2045-2046delAG测序图
对高通量测序的结果进一步分析发现,患儿LAMA2基因exon5测序深度较其他外显子低,推测患儿LAMA2基因exon5可能存在杂合缺失。选取ACTB基因为内参利用实时荧光定量PCR方法验证发现,相对于正常对照,患儿的exon5片段拷贝数降低了约50%,说明患者LAMA2基因exon5存在杂合缺失,且患者父亲亦存在该区域的杂合缺失(图3),说明患儿该区域的异常遗传自其父亲。

图3 实时荧光定量exon5拷贝数
2 讨论
CMD于1960年由日本学者福山首先报道。患儿出生时或出生后数月内即出现肌力和肌张力低下及关节挛缩,肌活检病理可见典型的肌营养不良改变,其他临床表现为脊柱后突、髋关节脱位、近端关节挛缩、斜颈等,后期运动能力变异很大,部分可以自由活动,而严重者始终不能独立行走[5]。该疾病主要分为福山型(Fukuyama type)和非福山型两大类,而非福山型又可分为merosin缺乏症(MDC1A)、肌-眼-脑病(又称Santavouri病)、Walker-Warburg综合征、先天性肌营养不良合并脊柱强直综合征以及Ullrich病等[3]。高通量测序技术又称下一代测序技术,能一次对几十万到几百万条DNA分子进行序列测定。国内外很多研究组利用该技术对相关疾病进行遗传学分析,如对杜氏/贝式肌营养不良、无脑回畸形及肥厚型心肌病等疾病的致病基因的检测[6-8],研究结果均显示高通量测序技术具有高通量、低成本以及高灵敏度的特点,适合致病基因不确定及基因较大疾病的检测。目前已经证实,多种基因突变与CMD发病相关[3],因此本例患儿选择与先天性肌营养不良相关的致病基因panel进行检测。
研究发现,LAMA2基因突变与MDC1A的发病有关。该基因定位于6q22-23区域,包括65个外显子,编码Laminin-a2蛋白(又称merosin)[9],基因突变的类型包括无义突变、错义突变、缺失重复以及剪接位点的突变,目前文献及数据库(HGMD database)中记录了约230多种LAMA2基因相关的突变。Merosin是一种糖蛋白,是异源三聚体复合物laminin-2和laminin-4的其中一条链,存在于横纹肌细胞外基质、神经-肌肉接头、皮肤以及滋养层等处,该蛋白参与细胞间的识别、细胞分化以及迁移等过程,对维持正常的血脑屏障功能、引导神经元移行有重要意义[10]。Merosin有6个结构域,与细胞外基质的大分子结合,建立一个连接肌细胞骨架和细胞外基质的桥梁。LAMA2基因突变后,merosin蛋白功能缺陷,使得肌膜完整性破坏,导致MDC1A发生[11]。
本例患儿胎儿期及出生时未见异常,出生后喂养困难,2岁可独坐,但至今不能独行。实验室检查发现肌酸激酶及其同工酶均升高,肌电图提示肌源性损害可能,磁共振显示脑白质异常信号。结合临床表现患儿疑似先天性肌营养不良。不足之处是本例患儿未作肌肉活检。基因检测发现患儿存在复杂杂合突变(c.2045-2046delAG,del Exon5),故患儿MDC1A诊断确立。通过文献搜索及数据库查询发现del Exon5为未见报道的新突变。
综上所述,MDC1A疾病起病较早,患儿出生时或出生后数月内即出现肌力和肌张力低下和关节挛缩,肌活检病理可见典型的肌营养不良改变。高通量测序可高效的进行遗传突变分析,有助于进一步明确诊断。
[1] Bönnemann CG, Wang CH, Quijano-Roy S, et al. Diagnostic approach to the congenital muscular dystrophies [J]. Neuromuscul Disord, 2014, 24(4):289-311.
[2] Muntoni F, Voit T. The congenital dystrophies in 2004: a century of exciting progress [J]. Neurmuscul Disord, 2004, 14(10):635-649.
[3] Falsaperla R, Praticò AD, Ruggieri M, et al. Congenital muscular dystrophy: from muscle to brain [J]. Ital J Pediatr, 2016, 42(1):78.
[4] 王硕,熊晖,罗静. 一个先天性肌营养不良1A型家系的临床、分子病理及遗传学研究 [J]. 中华医学遗传学杂志, 2010, 27(1):13-17.
[5] Ismail S, Schaffer AE, Rosti RO, et al. Novel mutation in the fukutin gene in an Egyptian family with Fukuyama congenital muscular dystrophy and microcephaly [J]. Gene, 2014, 539(2):279-282.
[6] Wang Y, Yang Y, Liu J, et al. Whole dystrophin gene analysis by next-generation sequencing: a comprehensive genetic diagnosis of Duchenne and Becker muscular dystrophy [J]. Mol Genet Genomics, 2014, 289(5):1013-1021.
[7] Jumuar SS, Lam AT, Kircher M, et al. Somatic mutations in cerebral cortical malformations [J]. N Engl J Med, 2014, 371(8):733-743.
[8] 刘旭霞,姜腾勇,朴春梅,等.目标基因捕获测序技术鉴定肥厚型心肌病致病突变的研究 [J]. 心肺血管病杂志, 2014, 33(4):599-603.
[9] Turner C, Mein R, Sharpe C, et al. Merosin-deficient congenital muscular dystrophy: a novel homozygous mutation in the laminin-2 gene [J]. J Clin Neurosci, 2015, 22(12):1983-1985.
[10] Kanagawa M, Toda T. The genetic and molecular basis of muscular dystriphy: roles of cell-matrix linkage in the pathogenesis [J]. J Hum Genet, 2006, 51(11):915-926.
[11] Holmberg J, Alajbegovic A, Gawlik KI, et al. Laminin-a2 chain-de fi ciency is associated with microRNA deregulation in skeletal muscle and plasma [J]. Front in Aging Neurosci, 2014, 6:155.
(本文编辑:蔡虹蔚)
Congenital muscular dystrophy type 1A: a report of one case with literature review
IANG Shiyuan,XIANG Na
(Department of Paediatrics,Shanxian Hygeia Hospital,Shanxian 274300,Shandong, China)
ObjectiveTo investigate the clinical features and genetic tests of a case with congenital muscular dystrophy type 1A (MDC1A).MethodsClinical data of proband were collected, and genetic change were tested using next generation sequencing, and literatures pertinent to the epidemiology, mechanisms, especially genetic testing of lisencephaly were reviewed.ResultsA 5 year and 2 month old boy present with normal intelligence and delayed motor development, he can be sit alone but not walk at two years old. Physical examination showed normal mental reaction, muscular dystrophy, hypotonia, and joint contracture at early age. From biochemical tests, we found creatine kinase (CK) and CK-MB were increased (491U/L, 41.8U/L). EMG test suggested possible muscle-derived damage. Brain MRI showed white matter abnormality. And a heterozygous mutation (c.2045-2046delAG) inherited from his mother in LAMA2 gene, and another novel heterozygous mutation (del Exon5) inherited from his father were identi fi ed by genetic test.ConclusionsLAMA2 gene de fi ciency can lead to MDC1A, and gene testing can help diagnosis.
congenital muscular dystrophy type 1A ; clinical features; genetic testing
10.3969/j.issn.1000-3606.2017.05.012
2016-11-01)
猜你喜欢
国际太空(2023年1期)2023-02-27 09:03:42
种子(2021年3期)2021-04-12 01:42:22
透析与人工器官(2020年1期)2020-11-16 01:42:34
铁道通信信号(2019年8期)2019-10-10 05:06:00
中国发展观察(2017年8期)2017-04-26 03:51:50
外语教学理论与实践(2016年1期)2016-06-11 05:51:48
新闻传播(2015年6期)2015-07-18 11:13:15
中国医疗美容(2015年4期)2015-04-27 02:24:06
中国医疗美容(2015年4期)2015-04-27 02:24:06
制造技术与机床(2015年10期)2015-04-09 07:06:10
