
EVALUATING DNA BARCODE MARKERS FOR FRESHWATER RED ALGAE: A CASE STUDY USING FAMILY BATRACHOSPERMACEAE
2017-05-16 09:07:50JILiFENGJiaNANFangRuCHENLeHUBianFangandXIEShuLian
水生生物学报 2017年3期
JI Li, FENG Jia, NAN Fang-Ru, CHEN Le, HU Bian-Fangand XIE Shu-Lian
(1. College of Environment and Safety, Taiyuan University of Science and Technology, Taiyuan 030024, China; 2. School of Life Science, Shanxi University, Taiyuan 030006, China; 3. Department of Biology, Jinzhong University, Jinzhong 030600, China)
EVALUATING DNA BARCODE MARKERS FOR FRESHWATER RED ALGAE: A CASE STUDY USING FAMILY BATRACHOSPERMACEAE
JI Li1, FENG Jia2, NAN Fang-Ru2, CHEN Le2, HU Bian-Fang3and XIE Shu-Lian2
(1. College of Environment and Safety, Taiyuan University of Science and Technology, Taiyuan 030024, China; 2. School of Life Science, Shanxi University, Taiyuan 030006, China; 3. Department of Biology, Jinzhong University, Jinzhong 030600, China)
DNA barcoding refers to the application of a small number of DNA fragments to achieve reliable, automatable species-level identification. In this study, the suitability of four candidate sequence regions were assessed-mitochondrial COI-5P and cox2-3 spacer, plastid rbcL and UPA-for species delimitation and discrimination in family Batrachospermaceae. The percentage of successful PCR amplifications of COI-5P, cox2-3 spacer, UPA, and rbcL markers was 96%, 100%, 96%, and 98%, respectively. COI-5P, UPA, and cox2-3 spacer sequence lengths were amenable to the acquisition of bidirectional sequencing reads using single primer pairs and met our size criterion of 300—800 bp. Phylogenetic analyses revealed that all four sequence regions were useful for species-level identification in the genus Batrachospermum except for some allied species. The two Chinese endemic species B. hongdongense and B. longipedicellatum were unable to differentiate from B. arcuatum using COI-5P, cox2-3 spacer, and rbcL markers, excepted for the UPA region. For species-level identification, the UPA locus exhibited the highest interspecific distances. We therefore recommended the plastid UPA gene as a standard DNA barcode in Batrachospermaceae, but acknowledge that there are no shared alleles between the endemic species.
DNA barcoding; Batrachospermaceae; Molecular phylogeny; Rhodophyta
DNA barcoding is the application of one or a few DNA fragments to achieve reliable, automatable identification at the species level[1,2]. The core idea behind DNA barcoding is the fact that sequence variation is ordinarily much lower among individuals than between closely related species. Following the initial assessment of the mitochondrial COI barcode in 2005, this diagnostic technology has attracted considerable attention as a powerful tool for algal species delimitation[3—16]. Remarkable progress has been achieved through the contributions of the large-scale DNA barcoding project and Red Algal Tree of Life initiatives[16].
A number of genomic regions used for phylogenetic analyses and species identification of algal samples over the past two decades have recently been investigated in greater detail for their suitability for barcoding analyses. Algal barcode genomic candidates have included chloroplast rbcL[14,17]and psbA[18], COI-5P[3,10,14]and cox2-3 spacer[18], nuclear SSU, LSU[10]and ITS[18,19], and plastid UPA[10,12,20,21]. The increasing number of publications had shown the superiority of COI-5P in species level identification for red macroalgae[3,10,14,21]. Domain V of the 23S plastid rRNA gene (UPA) as a DNA barcode also draws much attention in the identification of multiple eukaryotic algal groups. The UPA gene could be easily amplified and could distinguish samples at species level, even though the intra and inter species diversity values was relatively lower[12,22]. Many of thecited studies, however, have been based on incomplete sampling of large genera-thus overestimating the discriminatory power of the barcoding because an insufficient number of closely related species were considered—or have not entailed a comprehensive evaluation of potential barcode loci.
Batrachospermaceae, the largest freshwater red algal family in Rhodophyta, consists of nine genera comprising approximately 150 species[23]. Batrachospermum Roth, the type genus, has been split into Batrachospermum Roth and Kumanoa Entwisle, Vis, Chiasson, Necchi et Sherwood[23,24]based on molecular and morphological support, aiming reduce paraphyly with the Batrachospermum sensulato. For this case study, we selected four candidate sequence regions—COI-5P, cox2-3 spacer, UPA, and rbcL—to assess their suitability for species-level identification within Batrachospermaceae. The specific aims of this work were to evaluate ease of sequence acquisition and universality of primers, to analyze sequence variation and substitutional saturation at generaric level, and to estimate the effectiveness of these DNA barcodes for species differentiation within the genus.
1 Material and methods
1.1 Sampling strategy
In total, 49 samples representing four Batrachospermum, one Thorea, and one Bangia species were collected from China (Tab. 1). Two to seven individuals of each species were included. Voucher specimens were deposited at Shanxi University. Taxa, collection information, and DNA sequences were submitted to GenBank or the plant barcode data management system of Kunming Institute of Botany, Chinese Academy of Sciences. Specimens used for morphological examination were preserved in freshwater containing 4% formalin or 2.5% calcium carbonate-buffered glutaraldehyde, while those used for the molecular analysis were frozen at –20°C. To assess the utility of the four candidate markers for identification of these freshwater red algae, we additionally downloaded 214 sequences representing 11 species from GenBank.
1.2 DNA extraction, amplification, and sequencing
Total DNA was extracted using an Aqua-SPIN Plant gDNA Isolation mini kit (Watson Biotechnologies, Shanghai, China) following the manufacturer’s instructions. Primer pairs used to amplify the four regions (COI-5P, cox2-3 spacer, UPA, and rbcL) selected for barcoding Batrachospermum species were as follows: GazF1 and GazR1 (COI-5P[3]); cox2F and cox3R (cox2-3 spacer[25]); p23SrV_f1 and p23SrV_r1 (UPA[26]) and F160 and rbcL Rev (rbcL[26]) (Tab. 2). Standard polymerase chain reaction (PCR) amplifications were carried out in a MyCycler Thermal thermocycler (BIO-RAD, USA). PCR products were purified with a Gel Extraction mini kit (Watson Biotechnologies) according to the manufacturer’s recommendations for direct sequencing. The PCR products were sent to Takara Biotechnology Co. (Dalian, China) or Beijing AuGCT DNA-SYN Biotechnology Co. for sequencing.
1.3 DNA barcoding and phylogenetic analyses
Sequences were aligned and edited in ClustalX 2.0[27]. Pairwise Kimura 2-parameter distances were calculated for COI-5P, cox2-3 spacer, UPA and rbcL sequences in MEGA v4.1[28]to evaluate intraspecific and interspecific divergence of each candidate barcode. BLAST[29]was used to evaluate the genericlevel identification efficiency[30]of the four markers in the present study. Substitution saturation analyses for each marker were performed by using DAMBE v5.2.6[31].
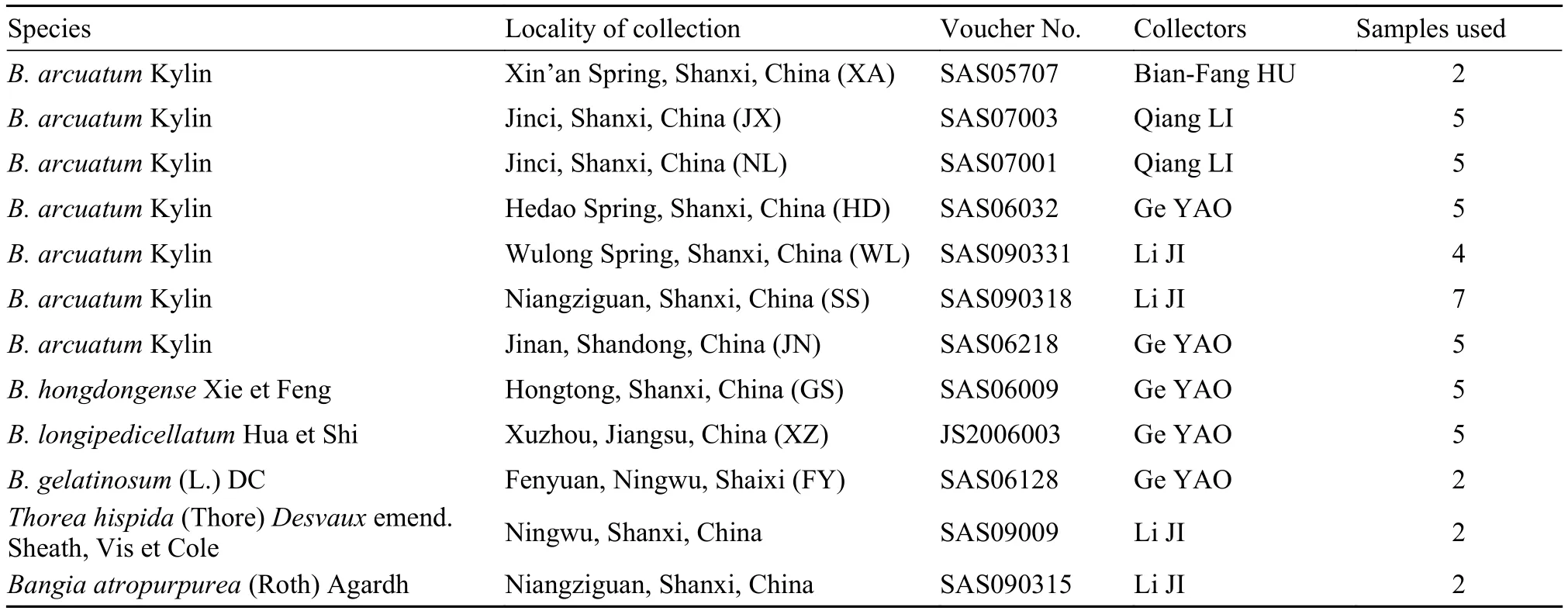
Tab. 1 Batrachopsermum and outgroup taxa sampled, collection, and voucher specimen information
Molecular identification and monophyletic assessment of species were performed in this study using two tree-based methods: maximum likelihood and Bayesian inference. The program jModeltest[32,33]was used to determine parameters for all maximum likelihood analyses, which were performed using PhyML 3.0[33]. Bootstrap resampling (1000 replicates) was carried out to estimate the robustness of trees generated from the maximum likelihood analysis[34]. Bayesian analyses were conducted in MrBayes 3.1.2[35]using a Metropolis-coupled Markov chain Monte Carlo algorithm running four simultaneous Markov chains. Each Markov chain was started from a random tree and run for 1000000 generations, sampling every 100 generations, for a total of 10000 samples per run. The first 2500 samples of each run were discarded as burn-in. The majority rule consensus tree was summarized from the remaining samples. Posterior probability was used to estimate robustness of Bayesian trees.
2 Results
2.1 Sequence analyses
The cited primers used in this study were universally applicable to all obtained samples, with target DNA regions successfully amplified and sequenced for most taxa (Tab. 3). The percentage of successful PCR amplifications of COI-5P, cox2-3 spacer, UPA, and rbcL regions was 96%, 100%, 96%, and 98%, respectively. Sequencing success rates were 86% for COI-5P, 100% for the cox2-3 spacer, 100% for UPA, and 92% for rbcL.
The length of the aligned COI-5P sequence dataset was 585 bp, with 236 (40%) informative sites. The aligned cox2-3 spacer dataset was 398 bp long and contained 182 (46%) informative sites. The length of aligned sequences in the UPA matrix was 338 bp, with 113 (33%) sites informative. A total of 115 rbcL sequences were generated; the aligned dataset encompassed 1124 bp including 419 (37%) informative sites. The percentage of indels in the tested loci ranged from 0 to 11.6% of the aligned sequences. Mean interspecific distances of the four target DNA regions were much greater than mean intraspecific distances, with the minimum interspecific distances of UPA and cox2-3 spacer sequences higher than those of the other two loci (Tab. 3). The distribution of intra- and interspecific distances is shown in Tab. 3.
We visually examined the transitional saturation of the four DNA fragments by plotting the estimated number of transitions and transversions for each pairwise comparison against the TN93 (Tamura & Nei distance) distance (Fig. 1). The transitions of the four DNA fragments have not achieved saturation, and the datasets can be used in phylogenetic analysis. For the complete COI-5P and UPA marker datasets, transitional saturation was reached at a distance of approximately 0.20 and 0.15, respectively. Saturation for cox2-3 spacer and rbcL markers occurred very close to their maximum divergence in these datasets.

Tab. 2 Primers used for PCR in the present study
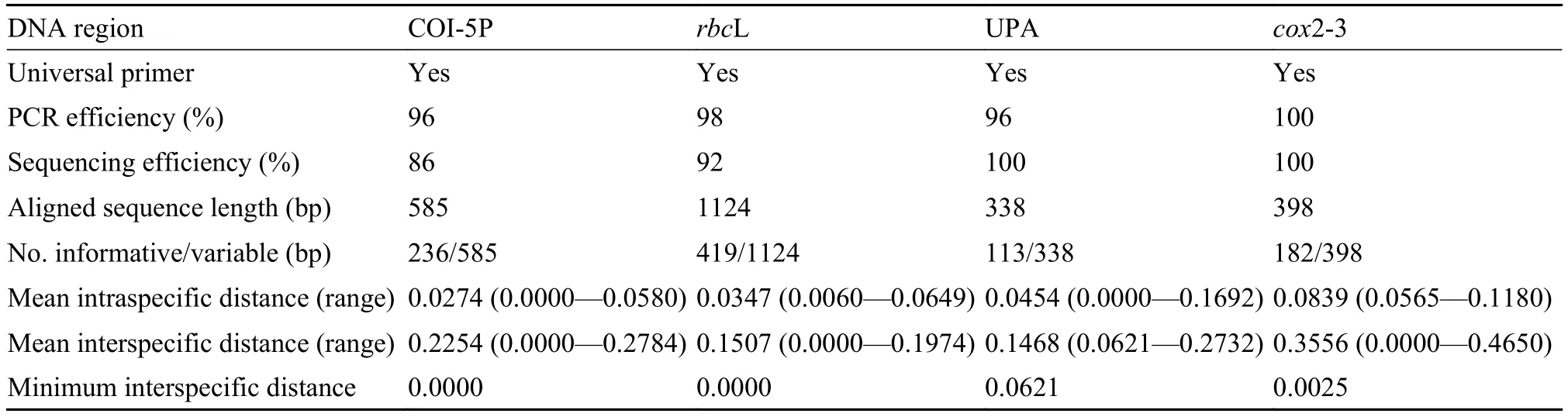
Tab. 3 Properties of the five candidate barcoding regions evaluated in the present study
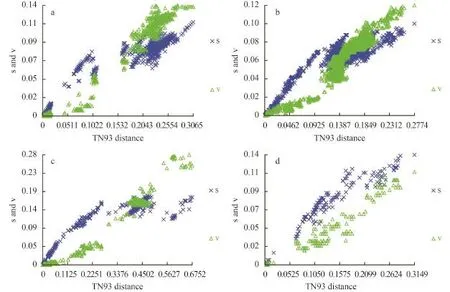
Fig. 1 Saturation curves for (a) COI-5P, (b) rbcL, (c) cox2-3 spacer, and (d) UPA markers for BatrachospermaceaeThe estimated number of transitions (indicated by ‘×’) and transversions (indicated by triangles) for each pairwise sequence comparison was plotted against TN93 distance
2.2 Assessment of monophyly
To test the ability of COI-5P, cox2-3, UPA, and rbcL markers to confirm the monophyly of each species, phylogenetic analyses based on maximum likelihood and Bayesian inference were carried out using sequences of all four barcode candidates. Because the topologies recovered by maximum likelihood and Bayesian analyses were similar, only the Bayesian trees are described. Support values for all analyses were shown as follows: Bayesian posterior probabilities/ ML bootstrap.
For phylogenetic analyses of COI-5P sequences, we used previously published data from nine Batrachospermum species: B. helminthosum Bory emend. Sheath, Vis et Cole, B. cayennense Montagne, B. guyanense (Montagne) Kumano, B. gelatinosum (Linnaeaus) De Candolle, B. arcuatum Kylin, B. hongdongense Xie et Feng, B. longipedicellatum Hua et Shi, B. turfosum Bory, and B. macrosporum Montagne. Thorea hispida (Thore) Desvaux emend. Sheath, Vis et Cole and Bangia atropurpurea (Roth) Agardh were selected as outgroups. Most terminal branches in the resulting Bayesian and maximum likelihood trees were strongly supported. Among the selected taxa, samples of six species formed well-supported monophyletic groups. B. hongdongense, B. longipedicellatum, and B. arcuatum from China, which are morphologically distinct, were closely associated with one another in COI-5P tree and therefore could not be distinguished solely on the basis of this DNA barcoding locus (Fig. 2). A similar relationship was observed among the three taxa in the rbcL trees (Fig. 3) and cox2-3 spacer trees (trees were not shown). In the UPA tree, however, a different placement was evident: B. hongdongense and B. longipedicellatum were separate from the B. arcuatum clade, but formed a separate clade sister with B. gelatinosum clade (Fig. 4).
To assess the suitability of the rbcL locus, we analyzed 11 Batrachospermum taxa: B. macrosporum, B. helminthosum, B. cayennense, B. turfosum, B. gelatinosum, B. arcuatum, B. hongdongense, B. longipedicellatum, B. pseudogelatinosum Entwisle et Vis, B. theaquum Skuja ex Entwisle et Foard, B. boryanum Sirodot, and B. antipodites Entwisle et Foard. Most of the taxa formed well-supported clades of distinct species. One of the five B. antipodites haplotypes (DQ523252), however, was included in the well-supported (100%) B. boryanum clade and may have been misidentified.
3 Discussion
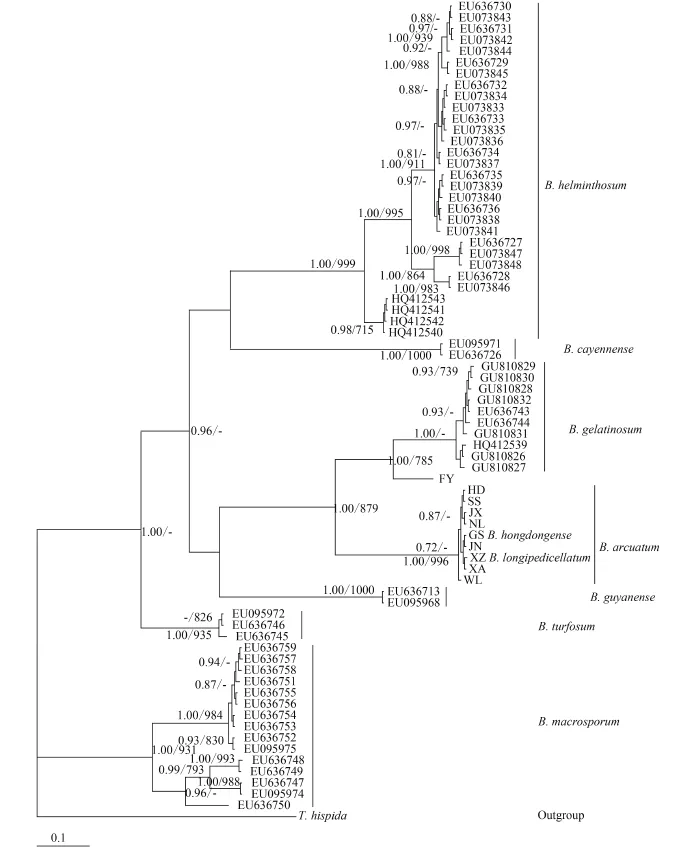
Fig. 2 Hypothesized phylogenetic relationships based on Bayesian analysis of the COI-5P maker for Batrachospermaceae specimensSupport values shown as Bayesian posterior probabilities / maximum likelihood (ML) bootstrap. Bootstrap values (>70%) are shown above the relevant branches. The same applies below
Four potential DNA barcode regions (COI-5P, cox2-3 spacer, UPA, and rbcL) were studied to assess their usefulness for species delimitation and discrimination within family Batrachospermaceae. The universality of PCR and sequencing primers is one of the most important criteria for candidate DNA barcoding markers[36]. In this study, primers to amplify the four candidate sequence regions performed well across all sampled species. The rbcL sequence primers have shown a high level of universality in freshwater red algae[26], working well for most Rhodophyta species, whereas degenerate primers were needed to amplify the cox2-3 spacer region.
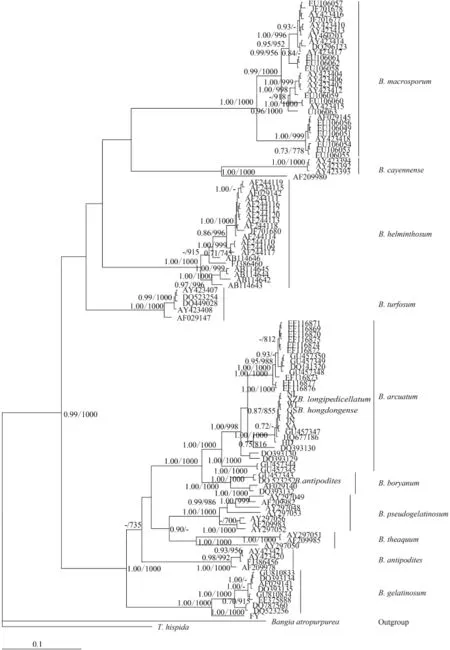
Fig. 3 Hypothesized phylogenetic relationships based on Bayesian analysis of the rbcL maker for Batrachospermaceae specimens

Fig. 4 Hypothesized phylogenetic relationships based on Bayesian analysis of the UPA maker for Batrachospermaceae specimens
Another criterion for an ideal DNA barcode is a relatively short sequence length (300—800 bp) to facilitate DNA extraction and amplification[1,2]. The lengths of the four proposed barcodes were given in Table 3. Sizes of COI-5P, UPA, and cox2-3 spacer sequences met these criteria and were amenable to the acquisition of bidirectional sequence reads using single primer pairs. The rbcL sequence can be used first because of the high amplification and sequencing success of its universal primer, while the cox2-3 spacer can be incorporated into analyses with ambiguous results or some cryptic species. Our data indicate that more sampling is needed to build a better picture of intraspecific variation. The UPA locus exhibited the highest interspecific distance for species-level identification and was more informative than the cox2-3 spacer sequence.
A portion of mitochondrial COI, namely, COI-5P, has been suggested for use as a barcode in the red algal group[3,5]. In this study, molecular analyses confirmed that COI-5P, rbcL, UPA, and cox2-3 spacer sequences work well for species-level identification of Batrachospermum except for some allied species. B. hongdongense and B. longipedicellatum, formerly placed in Batrachospermum sect. Batrachospermum and subsequently transferred into sect. Helminthoidea, are two Chinese endemic species that seem to have a closer relationship with B. arcuatum[24]. The results obtained for COI-5P, cox2-3 spacer, and rbcL sequences are in agreement with the findings of Ji et al.[24], who reported that 18S rDNA sequences of B. hongdongense and B. longipedicellatum were identical. In this study, COI-5P, rbcL and cox2-3 spacer sequences do not lend support for the classification of B. hongdongense, B. longipedicellatum, and B. arcuatum as distinct species as well, despite their clear morphological differences[37,38]. B. hongdongense was an endemic species, distinguished from other species based on carpogonial branches are long, straight, and not distinct from the primary fascicle cells[38]. B. longipedicellatum was first reported by Hua & Shi[37]which was also endemic to China. The distinct morphological features of B. longipedicellatum were that its branch not only arising from whorls, but also arising from cortex, carposporophyte obviously extending out of the whorl, trichogyne of carpogonia narrowly cylindrical, and branch also arising from the internode, which was very rare and only found in a few species[24]. However, the rbcL, COI-5P and cox2-3 spacer phylogenies resulted in identical topologies for B. hongdongense, B. longipedicellatum, and B. arcuatum which had not confirmed the attributes of the two endemic species proposed by the morphological features.
The UPA analysis presented here break up the phylogenetic framework of Batrachospermum hongdongense, B. longipedicellatum, and B. arcuatum. Compared with the other three potential DNA barcode regions, the UPA marker seems to provide more useful information and was able to separate B. hongdongense and B. longipedicellatum from the B. arcuatum branch, and formed a separate clade sister with B. gelatinosum clade. The high taxon sampling and more markers sequencing that represent the majority of type species for genera in the tribe may provide a strong phylogenetic framework in which taxonomy can be re-evaluated. Because B. hongdongense and B. longipedicellatum are both endemic to China, and can be clearly morphologically distinguished from varieties of B. arcuatum, it is practical to consider them as good species even in the absence of sufficient molecular evidence. For species-level identification, the UPA locus exhibited the highest interspecific distances in Batrachospermaceae. We therefore recommended the plastid UPA gene as a standard DNA barcode in Batrachospermaceae, but acknowledge that there are no shared alleles between the endemic species. A least, UPA gene has the potential to be an additional marker for COI barcode to
[1]Hebert P D N, Cywinska A, Ball S L, et al. Biological identifications through DNA barcodes [J]. Proceedings of the Royal Society of London B Biology, 2003, 270(1512): 313—321
[2]Hebert P D N, Ratnasingham S, Dewaard J R. Barcoding animal life: cytochrome c oxidase subunit 1 divergences among closely related species [J]. Proceedings of the Royal Society of London B Biology, 2003, 270(suppl. 1): 96—99
[3]Saunders G W. Applying DNA barcoding to red macroalgae: a preliminary appraisal holds promise for future applications [J]. Philosophical Transactions of the Royal Society of London Series B: Biological Sciences, 2005, 360(1462): 1879—1888
[4]Saunders G W. Routine DNA barcoding of Canadian Gracilariales (Rhodophyta) reveals the invasive species Gracilaria vermiculophylla in British Columbia [J]. Molecular Ecology Resources, 2009, 9(suppl. S1): 140—150
[5]Robba L, Russell S J, Barker G L, et al. Assessing the use of the mitochondrial cox1 marker for use in DNA barcoding of red algae (Rhodophyta) [J]. American Journal of Botany, 2006, 93(8): 1101—1108
[6]Lane C E, Lindstrom S, Saunders G W. A molecular assessment of northeast Pacific Alaria species (Laminariales, Phaeophyceae) with reference to the utility of DNA barcoding [J]. Molecular Phylogenetics and Evolution, 2007, 44(2): 634—648
[7]House D L, Vandenbroek A M, Vis M L. Intraspecific genetic variation of Batrachospermum gelatinosum (Batrachospermales, Rhodophyta) in eastern North America [J]. Phycologia, 2010, 49(5): 501—507
[8]Kucera H, Saunders G W. Assigning morphological variants of Fucus (Fucales, Phaeophyceae) in Canadian waters to reensure sufficient data, unless taxon-specific or more universal primer combinations for COI are designed, optimized and made available[10].
The utility of four short markers for construction of DNA barcode like data frameworks for a family of fresh water red algae have been contrasted in this paper. In the present study, the COI-5P, cox2-3 spacer and rbcL loci could identify most of the taxa correctly at species level in family Batrachospermaceae, but there were still a few species can not be identified or misidentified only through the candidate DNA barcode. Although only two endemic species were collected, it nonetheless indicates that the use of UPA marker should be expanded to a supplementary DNA barcode, at least in intractable taxa where routine DNA barcode have been found problematic.
Acknowledgements:
We thank Edanz Editing company for editorial assistance with the English.
[9]Sherwood A R, Vis M L, Entwisle T J, et al. Contrasting intra versus interspecies DNA sequence variation for representatives of the Batrachospermales (Rhodophyta): insights from a DNA barcoding approach [J]. Phycological Research, 2008, 56(4): 269—279
[10]Sherwood A R, Sauvage T, Kurihara A, et al. A comparative analysis of COI-5P, LSU and UPA maker data for the Hawaiian florideophyte Rhodophyta: implications for DNA barcoding of red algae [J]. Cryptogamie Algol, 2010, 31(4): 451—465
[11]McDevit D C, Saunders G W. On the utility of DNA barcoding for species differentiation among brown macroalgae (Phaeophyceae) including a novel extraction protocol [J]. Phycological Research, 2009, 57(2): 131—141
[12]Clarkston B E, Saunders G W. A comparison of two DNA barcode markers for species discrimination in the red algal family Kallymeniaceae (Gigartinales, Florideophyceae), with a description of Euthora timburtonii sp. nov [J]. Botany, 2010, 88(2): 119—131
[13]Le Gall L, Saunders G W. DNA barcoding is a powerful tool to uncover algal diversity: a case study of the Phyllophoraceae (Gigartinales, Rhodophyta) in the Canadian flora [J]. Journal of Phycology, 2010, 46(2): 374—389
[14]Manghisi A, Morabito M, Bertuccio C, et al. Is routine DNA barcoding an efficient tool to reveal introductions of alien macroalgae? A case study of Agardhiella subulata (Solieriaceae, Rhodophyta) in Cape Peloro lagoon (Sicily, Italy) [J]. Cryptogamie Algol, 2010, 31(4): 423—433
[15]Rueness J. DNA barcoding of select freshwater and marine red algae (Rhodophyta) [J]. Cryptogamie Algol, 2010, 31(4): 377—386
[16]Saunders G W, Moore T E. Refinements for the amplification and sequencing of red algal DNA barcode and RedToL phylogenetic markers: a summary of current primers, profiles and strategies [J]. Algae, 2013, 28(1): 31—43
[17]Geoffroy A, Le Gall L, Destombe C. Cryptic introduction of the red alga Polysiphonia morrowii Harvey (Rhodomelaceae, Rhodophyta) in the North Atlantic Ocean highlighted by a DNA barcoding approach [J]. Aquatic Botany, 2012, 100(1): 67—71
[18]Chong J, Jackson C, Kim J I, et al. Molecular markers from different genomic compartments reveal cryptic diversity within glaucophyte species [J]. Molecular Phylogenetics and Evolution, 2014, 76(1):181—188
[19]Ji L, Xie S L, Chen L, et al. Phylogeography of Batrachospermum arcuatum in North China based on ITS sequence data [J]. Chinese Journal of Oceanology and Limnology, 2014, 32(2): 372—376
[20]Nan F R, Feng J, Xie S L. Phylogenetic relationship of genus Kumanoa (Batrachospermuales, Rhodophyta) based on Chloroplast UPA genes [J]. Bulletin of Botanical Research,2014, 34(5): 584—591 [南芳茹, 冯佳, 谢树莲. 中国熊野藻属植物系统发育分析——基于叶绿体UPA序列. 植物研究, 2014, 34(5): 584—591]
[21]Zhao X, Pang S, Shan T, et al. Applications of three DNA barcodes in assorting intertidal red Macroalgal Flora in Qingdao, China [J]. Journal of Ocean University of China, 2013, 12(1): 139—145
[22]Sherwood A R, Presting G G. Universal primers amplify a 23s rDNA plastid marker in eukaryotic algae and cyanobacterial [J]. Journal of Phycology, 2007, 43(3): 605—608
[23]Entwisle T J, Vis M L, Chiasson W B, et al. Systematics of the Batrachospermales (Rhodophyta)-A synthesis [J]. Journal of Phycology, 2009, 45(3): 704—715
[24]Ji L, Xie S L, Feng J, et al. Molecular systematics of four endemic Batrachospermaceae (Rhodophyta) species in China with multilocus data [J]. Journal of Systematics and Evolution, 2014, 52(1): 92—100
[25]Zuccarello G C, Burger G, West J A, et al. A mitochondrial marker for red algal intraspecific relationships [J]. Molecular Ecology, 1999, 8(9): 1443—1447
[26]Vis M L, Sheath R G. A molecular investigation of the systematic relationship among Sirodotia species (Batrachospermales, Rhodophyta) in North America [J]. Phycologia, 1999, 38(4): 261—266
[27]Larkin M A, Blackshields G, Brown N P, et al. Clustal W and Clustal X, version 2.0 [J]. Bioinformatics, 2007, 23(21): 2947—2948
[28]Tamura K, Dudley J, Nei M, et al. MEGA4: Molecular evolutionary genetics analysis (MEGA) software version 4.0 [J]. Molecular Biology and Evolution, 2007, 24(8): 1596—1599
[29]Altschul S F, Gish W, Miller W, et al. Basic local alignment search tool [J]. Journal of Molecular Biology, 1990, 215(3): 403—410
[30]Little D P, Stevenson D W. A comparison of algorithms for the identification of specimens using DNA barcodes: examples from gymnosperms [J]. Cladistics, 2007, 23(1): 1—21
[31]Xia X. DAMBE v.5.2.6. Distributed by the author at http://dambe.bio.uottawa.ca. 2010
[32]Posada D. jModelTest: Phylogenetic Model Averaging [J]. Molecular Biology and Evolution, 2008, 25(7): 1253—1256
[33]Guindon S, Gascuel O. A simple, fast and accurate method to estimate large phylogenies by maximum-likelihood [J]. Systerms Biology, 2003, 52(5): 696—704
[34]Felsenstein J. Inferring phylogenies from protein sequences by parsimony, distance, and likelihood methods [J]. Methods in Enzymology, 1996, 266(1): 418—427
[35]Ronquist F, Huelsenbeck J P. MRBAYES 3: Bayesian phylogenetic inference under mixed models [J]. Bioinformatics, 2003, 19(12): 1572—1574
[36]Hollingsworth P M. DNA barcoding plants in biodiversity hot spots: progress and outstanding questions [J]. Heredity, 2008, 101(1): 1—2
[37]Hua D, Shi Z X. A new species of Batrachospermum from Jiangsu, China [J]. Acta Phytotaxonomica Sinica, 1996, 34(3): 324—326 [华栋, 施之新. 江苏串珠藻属一新种. 植物分类学报, 1996, 34(3): 324—326]
[38]Xie S L, Feng J. Batrachospermum hongdongense (sect. Batrachospermum, Batrachospermaceae), a new species from Shanxi, China [J]. Botanical Studies, 2007, 48(1): 459—464
cognized species using DNA barcoding [J]. Botany, 2008, 86(9): 1065—1079
DNA条形码在淡水红藻中的应用评价——基于串珠藻科植物
吉 莉1冯 佳2南方茹2陈 乐2胡变芳3谢树莲2
(1. 太原科技大学环境与安全学院,太原 030024; 2. 山西大学生命科学学院,太原 030006; 3. 晋中学院生物科学与技术学院,晋中 030600)
研究采用4种DNA序列, 分析了各片段序列特征以及在串珠藻科植物中种属水平的鉴定能力, 包括线粒体COI-5P、cox2-3 spacer序列, 以及叶绿体rbcL、UPA序列。结果表明, COI-5P、cox2-3 spacer、UPA以及rbcL序列的PCR扩增成功率分别为96%、100%、96%和98%。其中, COI-5P、cox2-3 spacer和UPA的片段大小符合标准DNA条形码的判定标准, 即片段大小在300—800 bp, 能够通过单对引物双向测序获得。系统发育分析的结果显示, 这4种DNA片段在串珠藻属植物的鉴定中能够鉴定大部分的种类, 但COI-5P、cox2-3 spacer以及rbcL序列均不能将两种中国特有种洪洞串珠藻B. hongdongense和长柄串珠藻B. longipedicellatum与弧形串珠藻B. arcuatum分开。在种水平的鉴定中, UPA基因的种间差异最大, 显示了较好的分离效果, 在串珠藻科植物的鉴定中可以作为一个标准的DNA条形码。
DNA条形码; 串珠藻科; 分子系统发育; 红藻门
Q949.29
A
1000-3207(2017)03-0643-09
10.7541/2017.82
Received date: 2016-06-06; Accepted date: 2016-11-12
Foundation item: Supported by the National Natural Science Foundation of China (31370239, 31440026); the PhD Start-up Fund of TYUST (20132013)
Brief introduction of author: JI Li(1982—), Female, Shanxi Linfen; Doctor; Research field: Phylogeny of fresh water red algae. E-mail: jili@tyust.edu.cn
XIE Shu-Lian, E-mail: xiesl@sxu.edu.cn, Fax: +86 (0)351 7018121
猜你喜欢
法医学杂志(2022年1期)2022-06-21 01:23:34
石材(2020年10期)2021-01-08 09:19:54
核农学报(2019年1期)2019-01-09 08:04:44
中国环境科学(2018年7期)2018-07-26 09:03:50
创新作文(1-2年级)(2017年10期)2018-04-16 10:17:36
科学家(2017年23期)2018-01-11 20:54:58
数学小灵通(1-2年级)(2017年5期)2017-06-05 09:12:14
创新作文(小学版)(2017年28期)2017-03-21 02:54:25
法医学杂志(2016年5期)2016-11-21 01:44:43
中国环境科学(2015年6期)2015-11-19 08:39:58
