
卤代金刚烷衍生物的合成研究进展
2017-05-07 03:35:01刘田英曹端林王艳红王建龙李永祥
含能材料 2017年1期
刘田英, 曹端林, 王艳红, 王建龙, 李永祥, 罗 军
(1. 中北大学化工与环境学院,山西 太原 030051; 2. 南京理工大学化工学院, 江苏 南京 210094)
1 引 言
有机笼状高能化合物因其结构紧凑,对称性高,在医药、高分子、含能材料等有机化合物中具有重要的应用价值,主要包括八硝基立方烷(ONC)、六硝基六氮杂异伍兹烷(CL-20)和多硝基金刚烷(PNAs)等[1]物质。目前ONC在国外只限于实验室合成,而国内还未见合成报道[2]; CL-20产量目前国内已达到吨级每年[3]; 而PNAs的合成也是国内外研究的热点。以金刚烷为母体合成的一系列卤代金刚烷衍生物,是合成PNAs重要的有机中间体。
金刚烷(1)又名三环[3.3.1.1]癸烷(Fig.1),分子式C10H16,是一种高度对称的笼状烃,密度大、热值高、良好的热稳定性、润滑性和亲油性[4]。诺贝尔化学奖获得者Olah[5]曾预言,随着金刚烷结构性能及规律性联系的研究日趋深入和系统化,相关的生产规模和应用领域也将急剧扩大,未来将出现一门新的科学—“金刚烷化学”。金刚烷化学有可能改变高密度、高能量燃料的生产方法,为航空航天及军事领域提供新型化学推进剂[6],其衍生物的合成研究将成为今后的研究重点。

图1 金刚烷结构式[4]
Fig.1 The molecular structure of adamantane[4]
金刚烷分子中的氢原子可以同时或分别被卤素原子取代,生成一系列的卤代金刚烷衍生物。卤代金刚烷衍生物作为金刚烷衍生物中重要的一类化合物,已经在生物医药、功能高分子、润滑油、催化剂、表面活性剂、感光材料、农药等众多领域广泛应用,被称为新一代精细化工原料[7-8]。特别是在含能材料领域,以卤代金刚烷衍生物为前体合成的多硝基金刚烷,稳定性好,具有较高的能量,而且对热和撞击非常钝感,是近年来很受推崇的高能量密度材料[9]。本文参考国内外文献,综述了以金刚烷为“母体”的卤代金刚烷衍生物的合成方法,着重对卤代金刚烷中的分离问题进行了简要评述,为以卤代金刚烷为中间体研发新型的金刚烷衍生物提供参考。
2 卤代金刚烷衍生物
2.1 卤代金刚烷的合成机理
金刚烷分子中1、3、5、7位4个叔碳原子上的氢原子较其他仲碳原子上的氢原子易发生取代反应,其中最典型的是 SE2 型亲电取代反应和 SN1 型亲核取代反应,由三个不同空间角度的六元环结构组成的金刚烷,其桥碳原子及与其直接相连的三个原子不处在同一平面,不易实现sp2杂化,降低反应的活性,而金刚烷高度对称的结构使得桥碳原子与相连的三个原子能够形成独特的半平面结构[10],使sp2杂化成为可能,从而在一定的条件下发生取代反应。金刚烷桥碳原子的氢活化取代机理可以运用 SET(Single-Electron Transfer)理论解释[11],其过程如下所示:
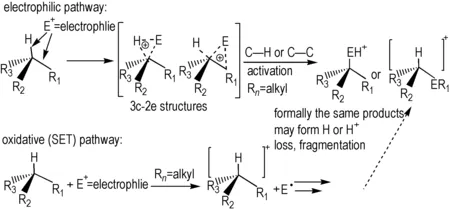
Scheme 1 Activation mechanism of bridge carbon atoms[11]
对于金刚烷桥碳原子的氢原子取代可以用Scheme 2的两个独立过程分别描述,其中Scheme 2a为亲电取代过程,Scheme 2b为SET氧化过程,两种过程都能很成功的解释其取代机理[12]。
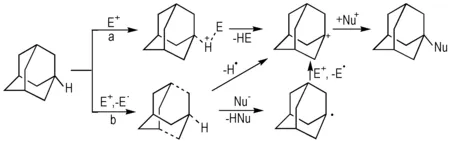
Scheme 2 Replace mechanism of adamantane bridge carbon atoms[12]
2.2 卤代金刚烷的合成及应用
2.2.1 氟代金刚烷
氟代金刚烷在PET成像应用程序和医药等领域有着重大的潜在价值,在金刚烷分子中引入氟原子可以增加金刚烷的活性,常用的氟化试剂有二氟化氙、1-氯甲基-4-氟-1,4-二氮杂双环[2.2.2]辛烷二(四氟硼酸)盐(Selectfluor氟试剂)、氟化银等。
2.2.1.1 1-氟金刚烷
1-氟金刚烷(3)主要是以1-金刚烷甲酸(2)、金刚烷(1)为原料f化学法得到。
(1)以1-金刚烷甲酸(2)为原料
由2可以通过不同的氟化试剂反应得到3。1986年,Patrick等[13]以二氟化氙为氟化试剂合成3(Scheme 3a); 该法是将2和二氟化氙在二氯甲烷溶剂中,22 ℃下反应10 h,合成3,产率为82%。2012年,Yin等[14]以Selectfluor氟试剂为氟化试剂(Scheme 3b),2为原料,硝酸银为催化剂,合成3; 分别对比了在以丙酮/水为混合溶剂和水为单一溶剂,在以丙酮/水为溶剂时,产率为22%,而以水为溶剂时产率可达67%。2015年,Huang等[15]以Et3N-3HF为氟化试剂,[Mn(tmp)]Cl为催化剂,合成3,产率为38%(Scheme 3c)。
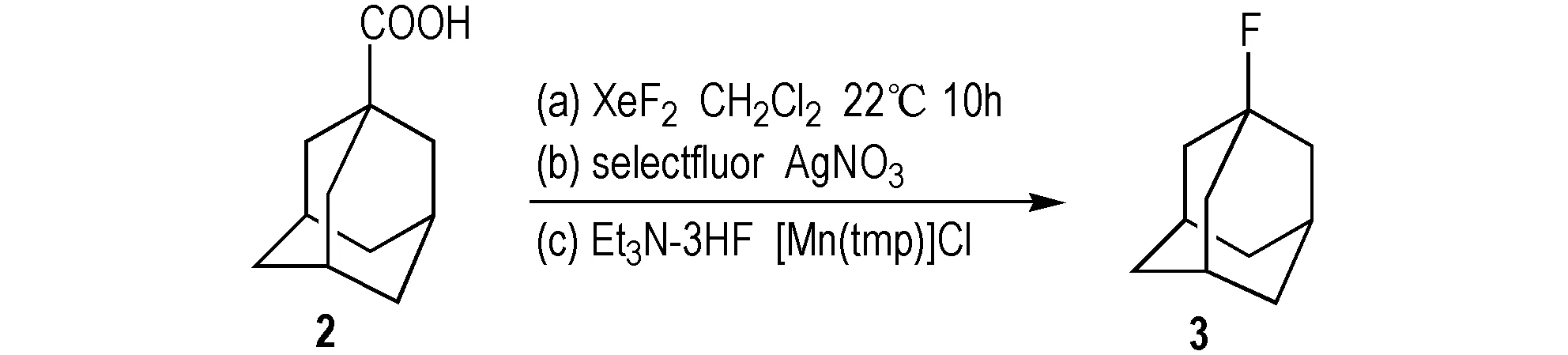
Scheme 3 Synthetic route of 1-fluoroadamantane by adamantane-1- carboxylic acid[13-15]
上述路线中,用二氟化氙作为氟化试剂,产率较高,但因其具有的亲电性和高活性,所合成的官能团表现出较差的耐受性和较低的选择性,使得这类反应的应用受到很大的局限; 用Selectfluor氟试剂为氟化试剂,水做溶剂,副产物少,提纯分离工艺较简单; 用Et3N-3HF为氟化试剂,虽然产率较低,但反应条件温和,操作简单。
(2)以金刚烷(1)为原料
2012年,Liu等[16]以1为原料,[Mn(tmp)]Cl为金属催化剂,合成3(Scheme 4); 该法是1和氟化银在催化剂下50 ℃反应8 h,并通入氮气保护,合成3,产物中同时存在3和2-氟金刚烷(4)两种物质,总产率为53%。该催化剂催化活性高,反应条件较温和,而且催化剂可以重复使用,成本低。
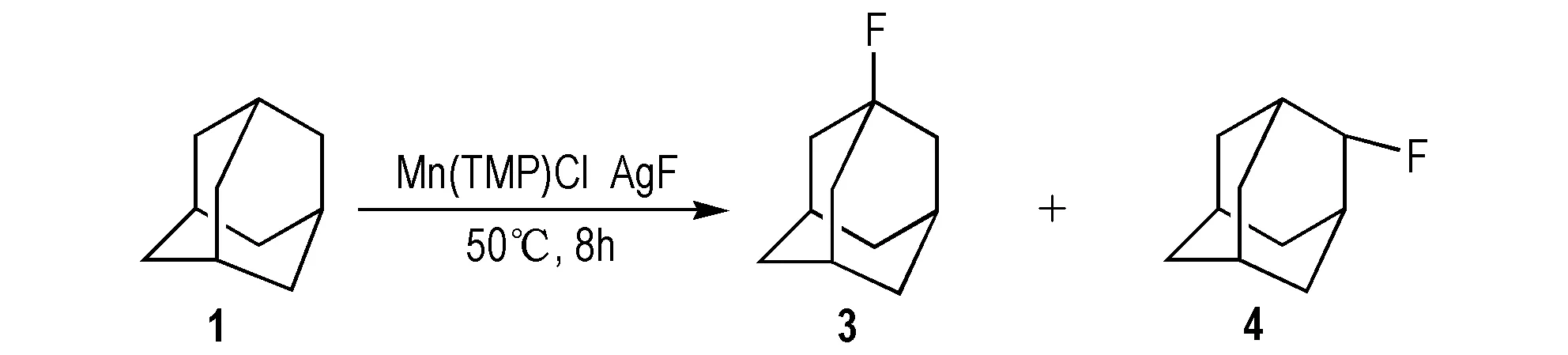
Scheme 4 Synthetic route of 1-fluoroadamantane and 2- fluoroadamantane by adamantane[16]
2014年,Pitts等[17]以1为原料,2-苯甲基环己酮为非金属催化剂,合成3(Scheme 5); 该法是将1、Selectfluor氟试剂和2-苯甲基环己酮按一定比例加入圆底烧瓶,通入氮气保护,缓慢滴加乙腈,室温反应4 h,合成3,产率为85%。该法催化剂选择性高,毒性小,实验操作简单,但催化剂不易得。

Scheme 5 Synthetic route of 1-fluoroadamantane by adamantane[17]
(3)电化学法
传统的氟代反应中存在一定的局限性,常无法在特定碳位置引入相应的氟原子。2008年,Aoyama等[18]通过电化学氟化的方法合成3(Scheme 6); 该法是以1为原料,Et3N-5HF为氟化试剂,和二氯甲烷在35 ℃,2.3 V电压下发生氟化作用,合成3。实验中随着电压升高,氟代金刚烷中氟原子个数增加,当电压增至60 V时,产物中出现1,3,5,7-四氟金刚烷(7)。用电化学法制备3,对实验仪器的依赖性较强,试剂昂贵不易得,合成产物为3、3,5-二氟金刚烷(5)、1,3,5-三氟金刚烷(6)、1,3,5,7-四氟金刚烷(7)的混合物,由于金刚烷结构的高度对称性,各产物之间极性相差很小,分离存在一定的难度,但其氧化电势的控制简单精准,反应条件温和,可以通过控制不同的电压,在相应的位置引入氟原子,采用电化学的方法为合成氟代金刚烷提供了一种新的思路,在有机合成中有着重要的意义。
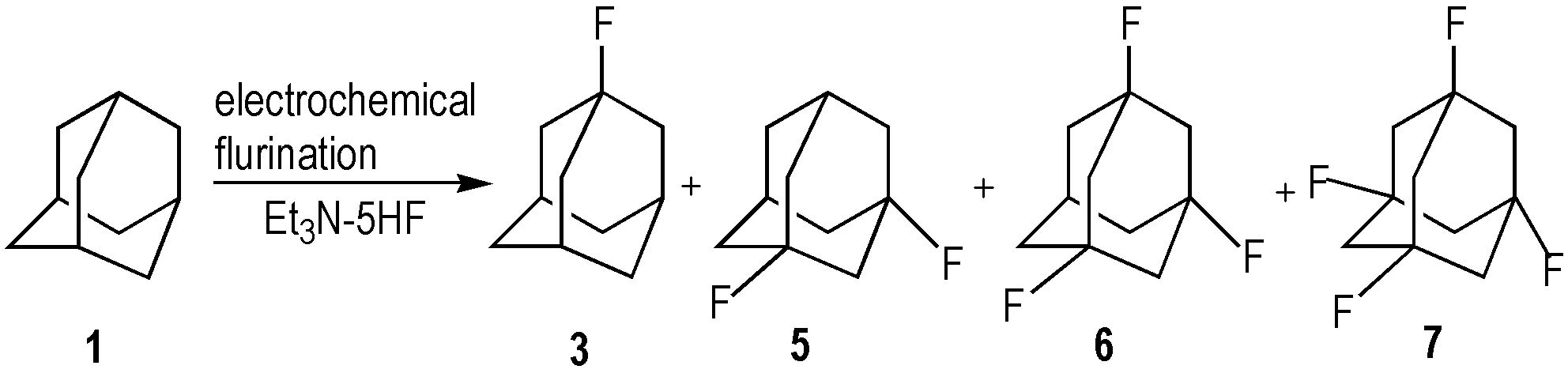
Scheme 6 Electrochemical preparation of 1-fluoroadamantane[18]
2.2.1.2 1,3,5,7-四氟金刚烷
1,3,5,7-四氟金刚烷(7)是重要的金刚烷衍生物,四个稳定的C—F键可以使四个桥头碳更稳定,从而成为合成桥碳金刚烷衍生物的重要有机中间体。
2013年,易文斌等[19]以1为原料,三氯化铝为催化剂合成7(Scheme 7)。具体方法为: Selectfluor氟试剂、三氯化铝催化剂和1,在溶剂为甲醇和水的混合溶液中,10~50 ℃下反应,合成7,产率为72%。该方法有以下优点: 使用的氟化试剂是新型的固体氟化试剂,便于储存使用,无毒无污染,反应条件温和,无需高温高压,无需耐腐蚀反应容器,无需特殊反应仪器,操作简单。

Scheme 7 Synthetic route of 1,3,5,7-fluororoadamantane[19]
2.2.2 氯代金刚烷
氯代金刚烷的制备主要通过金刚烷与各种氯化剂反应制得,常用的氯化剂有氯气、次氯酸钠、氯化亚砜和氯磺酸等,由于金刚烷结构的高度对称性,反应所得产物一般为各种氯代金刚烷的混合物。
2.2.2.1 1-氯金刚烷
1-氯金刚烷(8)可由1为原料,通过不同的氯化剂反应合成。1961年,Smith等[20]以氯气为氯化剂,1为原料,四氯化碳为溶剂,25~30 ℃下光照反应2 min,合成8(Scheme 8a)。1971年,Petter等[21]以次氯酸叔丁基酯为氯化剂,1为原料,三氯化铝为催化剂,合成8(Scheme 8b),产率为89.9%。2015年,Draksharapu等[22]以次氯酸钠为氯化剂,1为原料,镍络合物为催化剂,二氯甲烷为溶剂,20 ℃下反应2 h,合成8(Scheme 8c),产率为11.7%。

Scheme 8 Synthetic route of 1-chloroadamantane by adamantane[20-22]
上述氯化剂的选择中,氯气的毒性大,选择性较差,光催化反应对实验仪器有一定的依赖性,但氯化剂活性高,大大缩短了反应时间; 次氯酸叔丁基酯选择性较好,产率较高,但后处理步骤较繁琐; 次氯酸钠为氯化剂,虽产率较低,但其镍络合物催化剂催化活性好、机械强度高、对毒物不敏感、导热性好。
2012年,Wang等[23]以2为原料,采用一价银与1,10-啡咯啉形成的络合物Ag(Phen)2OTf为新型催化剂,合成8(Scheme 9); 该法是在惰性气体的保护下,根据Hunsdiecker反应,将2和次氯酸叔丁基酯溶解在乙腈中,加入催化剂,20 ℃下反应3 h,得到8,产率为93%; 该法反应条件温和、催化剂催化活性高,具有较好的化学选择性和官能团兼容性,绿色环保,在有机合成中有着良好的应用前景。
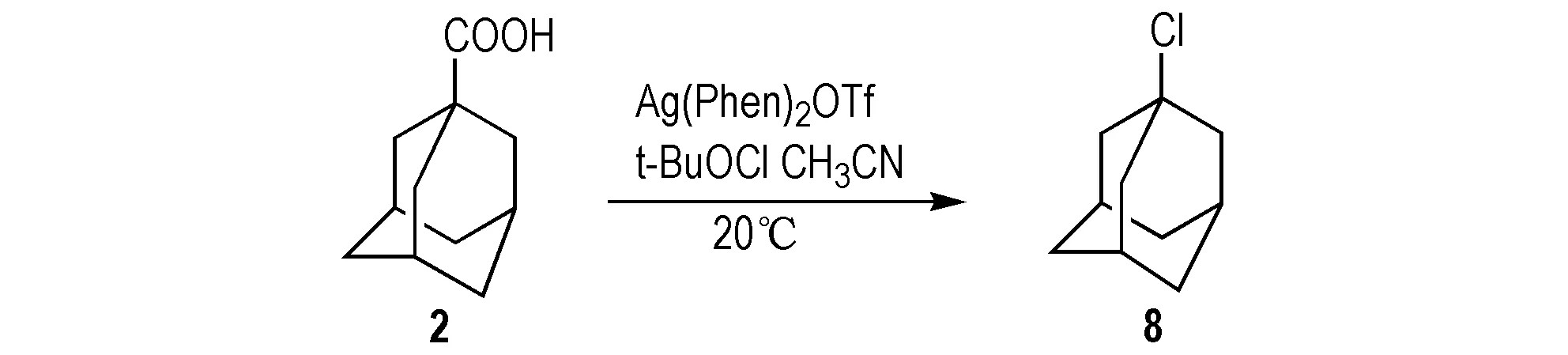
Scheme 9 Synthetic route of 1-chloroadamantane by adamantane-1-carboxylic acid[23]
2013年,Mondal等[24]以9为原料,二氯亚砜为氯化剂合成8(Scheme 10); 该法是将9和二氯亚砜加入二氯甲烷中,0 ℃下反应1 h,金刚烷上的醇羟基被置换为磺酰氯基团,生成1-磺酰氯金刚烷(10),之后以四氯化钛为催化剂进一步反应得到8,产率为92%。
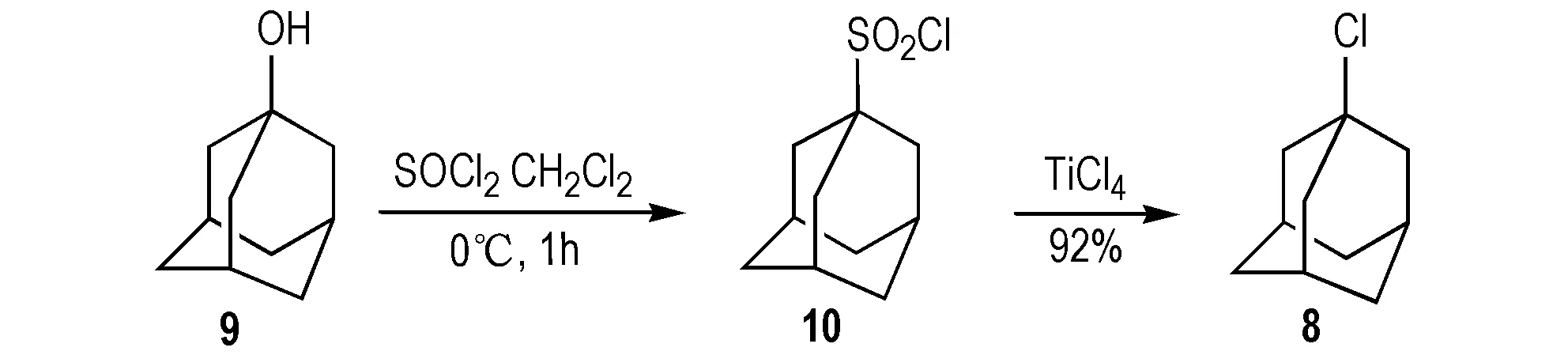
Scheme 10 Synthetic route of 1-chloroadamantane by 1-adamantanol[24]
2.2.2.2 1,3,5,7-四氯金刚烷
金刚烷的桥头碳原子特别活泼,容易发生亲核取代反应。目前文献报道合成1,3,5,7-四氯金刚烷(11)的方法通常都是由1和三氯化铝催化剂在不同的反应条件下得到。
2004年,Lee等[25]以三氯化铝作为催化剂,四氯化碳为溶剂,加热反应1 h,合成化合物11。Lee通过改变催化剂与原料的比例大大缩短了反应时间,提高了产率,产率为98%。2014年,Singh等[26]在Lee的基础上,室温下反应合成11(Scheme11),该反应条件更加温和,产率为91%。2014年,杜耀等[1]在Lee的基础上研究了不同温度、反应时间对氯代反应的影响,得出最佳的反应时间为3 h,温度为70 ℃。但是所得产物中同时存在三氯代金刚烷以及五氯代金刚烷,根据氯代金刚烷易升华的特性,对其进行提纯分离,由于同分异构体的存在,产物并未能完全分离,其多氯代金刚烷的提纯分离工艺仍需进一步研究。
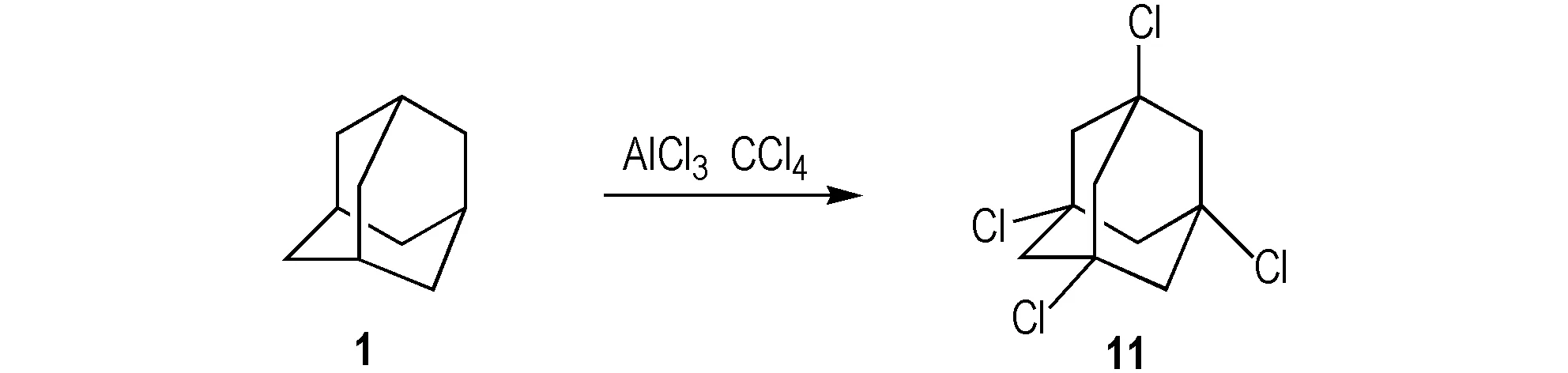
Scheme 11 Synthetic route of 1,3,5,7-tetrachloroadamantane[26]
Khusnutdinov等[27]改变了传统的催化剂,以1为原料,锰为催化剂,四氯化碳为溶剂,合成11(Scheme 12)。实验研究了用钼,钒,锰等不同的催化剂对氯代反应的影响,得出最佳的催化剂为锰,但所得产物中同时含有11,1,5-二氯金刚烷(12),1,3,5-三氯金刚烷(13)三种物质,其分离提纯问题仍需进一步研究。
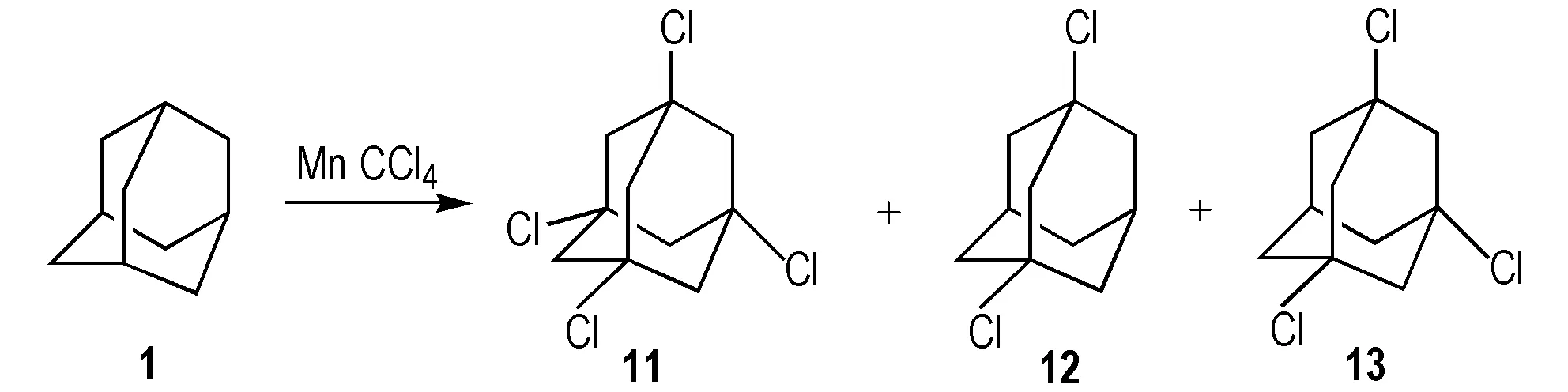
Scheme 12 Synthetic route of 1,3,5,7-tetrachloroadamantane[27]
由于金刚烷结构的高度对称性,反应所得产物一般为各氯代金刚烷混合物,各产物之间极性相差较小,在除水之外的溶剂中溶解度都很相近,目前尚未找到合适的展开剂和溶剂,通过薄层色谱和重结晶等方法进行分离,对于多氯代金刚烷的分离提纯工艺将会是今后研究的重点。
2.2.2.3 多氯代金刚烷甲酸
多氯代金刚烷甲酸作为合成多硝基金刚烷衍生物的重要有机中间体,在含能材料等领域有着广泛的应用,其合成研究具有重要意义。
(1)3-氯金刚烷甲酸
Yurchenko等[28]以2为原料,四氯化碳为溶剂和一氯化碘加热反应5 h,合成3-氯金刚烷甲酸(14)(Scheme 13a),产率为85%。2004年,Khusnutdinov等[27]以2为原料,乙酰丙酮锰作为催化剂,四氯化碳为氯化剂,在乙腈溶液中,200 ℃下反应1 h,合成14(Scheme 13b),产率为85%; 该法中乙腈的使用量较大,但催化剂活性高,而且可以重复使用,大大缩短了反应时间短。
(2)3,5-二氯金刚烷甲酸
专利WO2007/140439A2[29]报道了以二氯化钌为催化剂,合成3,5-二氯金刚烷甲酸(化合物15)的方法(Scheme 13c); 该法是将2溶解在乙酸乙酯中,加入二氯化钌、水、一定量的次氯酸钠,80 ℃下反应12 h,合成15。
(2)八氯代金刚烷甲酸
金刚烷常规的氯代反应,一般情况下,最多可以在分子中引入4个氯原子,如采用光催化反应,可以在分子中引入更多的氯原子。Marvin[30]在专利US3218155中报道了八氯代金刚烷甲酸(化合物16)的合成方法(Scheme 14); 该法是以2为原料,四氯化碳为溶剂,紫外光照下通入氯气合成16。但专利中未对产物进行表征分析,且未给出具体产物16的结构式,其16中氯所在的具体位置也尚未给出。光催化反应制备多氯代金刚烷甲酸反应属于自由基亲核取代反应,其3、5、7 位三个桥头叔碳原子上的氢原子具有较强的化学反应活性,更容易被氯原子取代。近年来,国内的学者没有对该物质进一步的研究,本课题组在它的合成工艺方面进行了研究,目前已经取得了一定的进展[1]。光催化反应制备多氯代金刚烷甲酸有着以下优点: 时间短、催化活性高、转化效率高、实验操作方法简单、温度低、成本低,符合现代绿色化学的需求。

Scheme 13 Synthetic route of 3,5-dichloroadamantane-1-carboxylic acid and 3-chloroadamantane-1-carboxylic[27-29]

Scheme 14 Synthetic route of octchloroadamantane-1-carboxylic acid[30]
2.2.3 溴代金刚烷
溴代金刚烷作为合成金刚烷胺等化合物的有机中间体,在抗病毒等医药领域有着广泛的应用价值,其合成主要通过1与液溴反应制得,通过改变催化剂或反应物料比,可以选择性制备含有不同溴原子数目的金刚烷桥头碳取代物。常用的溴化剂有二溴甲烷、溴水等,但溴水、二溴甲烷对实验仪器腐蚀较大,高温下产生的溴化氢气体对环境污染较大,不能大批量生产,因此寻找新的溴化试剂和新的合成路线显的尤为重要。
2.2.3.1 1-溴金刚烷
1-溴金刚烷(17)是重要的有机合成中间体,由它可以合成一系列衍生物。金刚烷的一溴代反应是自由基反应,光、热或自由基引发剂(如过氧化物等)是促进反应进行的必要条件,将金刚烷溶于液溴加热反应一定时间可以得到17。
1988年,Cyril等[31]等以1为原料和液溴反应合成17,实验中所需的液溴需进行提纯干燥,该法虽制备反应条件比较苛刻,但克服了高温反应中液溴挥发的问题,使反应条件更温和。
2013年,Nishina等[32]以Li2MnO3为催化剂制备17; 该法是将1溶解在溴水里,加入催化剂,光照下100 ℃反应10 h,所得产物同时存在17和2-溴金刚烷(18)(Scheme 15)。该催化反应催化活性高,绿色无污染,而且可以重复使用,是溴代反应历程中一个新的突破。
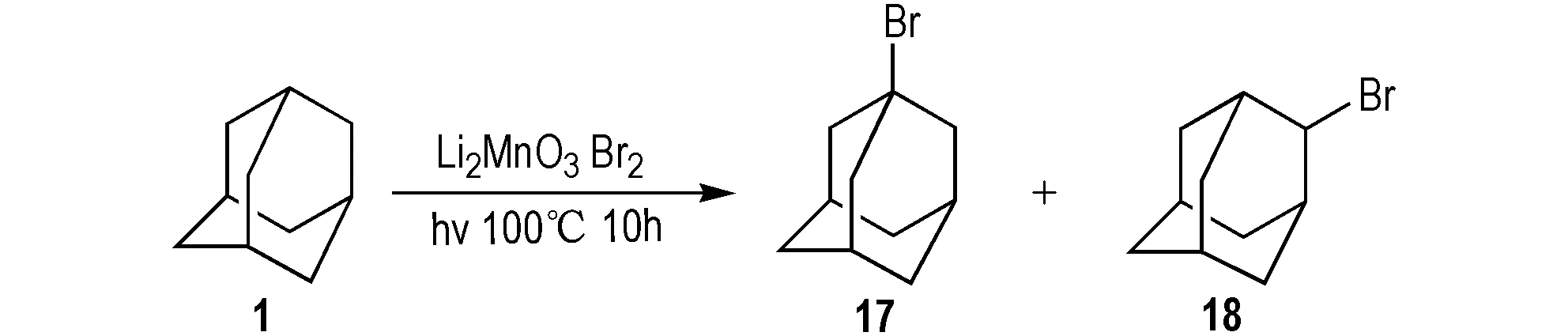
Scheme 15 Synthetic route of 1-bromoadamantane and 2-bromoadamantane[32]
2015年,Khusnutdinov等[33]以1为原料,Fe(acac)3为催化剂,四溴化碳为溴化剂,正己烷为溶剂,80 ℃反应2 h得到17(Scheme 16a)。实验研究了不同催化剂Fe2(CO)9、Fe3(CO)12、Fe(acac)2、Fe(acac)3、 Fe(OAc)2·4H2O、FeCl3·6H2O、Fe(C8H15O2)3对溴代反应的影响,最佳的催化剂为Fe(acac)3。用Fe(acac)3作催化剂,产物中同时存在17和1,3-二溴金刚烷(19)。Schreiner等[34]同样以1为原料,苯基三甲基铵为相转移催化剂,在50%氢氧化钠和四溴化碳的混合溶剂中,同样得到产物17和19,其中17占70%,19占15%(Scheme 16b)。

Scheme 16 Synthetic route of 1-bromoadamantane and 1,3-dibromoadamantane[33-34]
上述Scheme 14b中以四溴化碳为溴化试剂碳原子的活化机理可以用过程Fig.2来解释: 其中E1,E2符合所Fig.2中的SET 反应机理,而 E3,E4则符合自由基取代机理。虽然1-,2-金刚烷自由基具有相似的热力学稳定性[35],但是由于空间位阻效应,在 E3,E4两步反应中的氢吸引和自由基取代过程中,使得 2-金刚烷取代产物量很少。
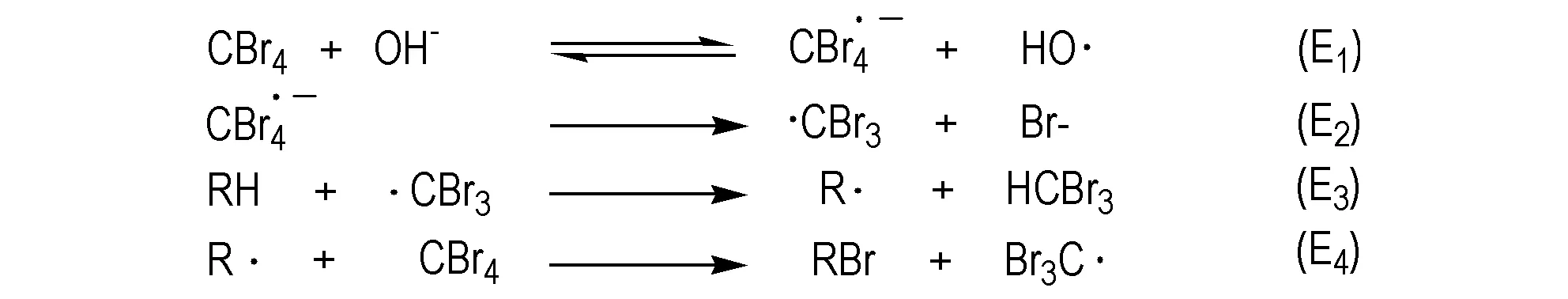
图2 碳原子的活化机理[12]
Fig.2 Activation mechanism of carbon atoms[12]
Kawakami等[36]以9为原料制备17(Scheme 17); 该法是将9溶解在二溴甲烷中,室温回流反应10 h,可得17,产率为99%。该法反应条件温和、产率高、无需催化剂、成本低、操作简单。

Scheme 17 Synthetic route of 1-bromoadamantane by 1-adamantanol[36]
2013年,Inomata等[37]以1-丁氧基金刚烷(20)为原料合成17(Scheme 18); 该法是将溴水加入17中,并加入四氯化碳,室温搅拌反应12 h,合成17,产率为97%。
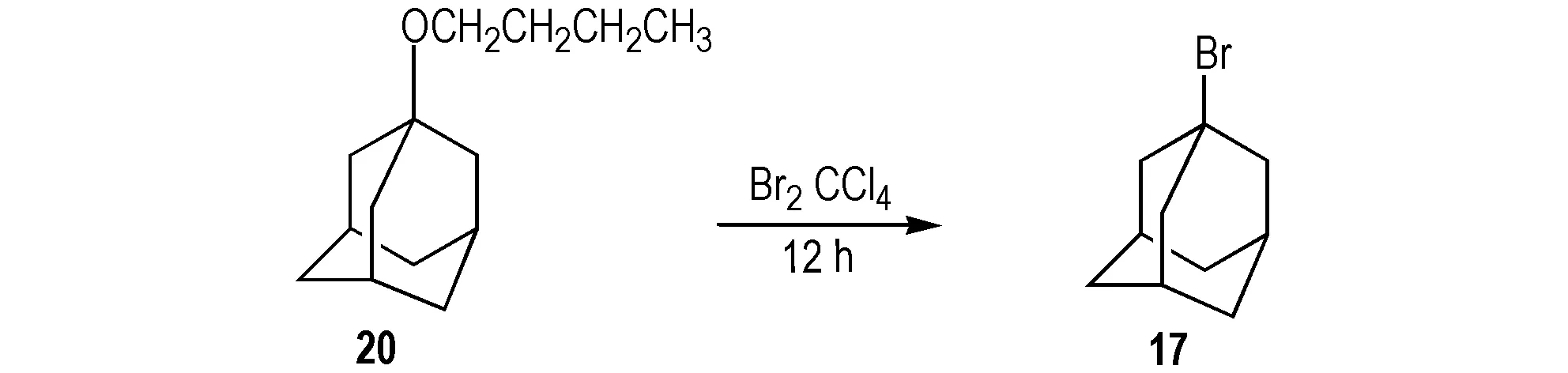
Scheme 18 Synthetic route of 1-bromoadamantane by 1-butoxyadaman[37]
2015年,Mizukami等[38]以3为原料合成17(Scheme 19); 该法是将3、二溴甲烷和三乙基铝在20 ℃下反应0.5 h,产率为80%。该反应条件温和,时间短,但原料3价格昂贵不易得,合成较困难,不能大量生产。

Scheme 19 Synthetic route of 1-bromoadamantane by 1-flouroadamantane[38]
2.2.3.2 二溴代金刚烷
二溴代金刚烷主要作为合成对桥头二取代金刚烷衍生物的中间体,可由1和溴化试剂在三氯化铝、铑、铁等的催化剂下反应制得。
1991年,Denmark等[39]以三氯化铝为催化剂两步法合成19; 该法先以1为原料,和溴水反应得到化合物17,再以三溴化铝为催化剂,三溴化硼为溴化试剂,85 ℃下和溴水反应1.5 h,得到19,产率为87%(Scheme 20)。
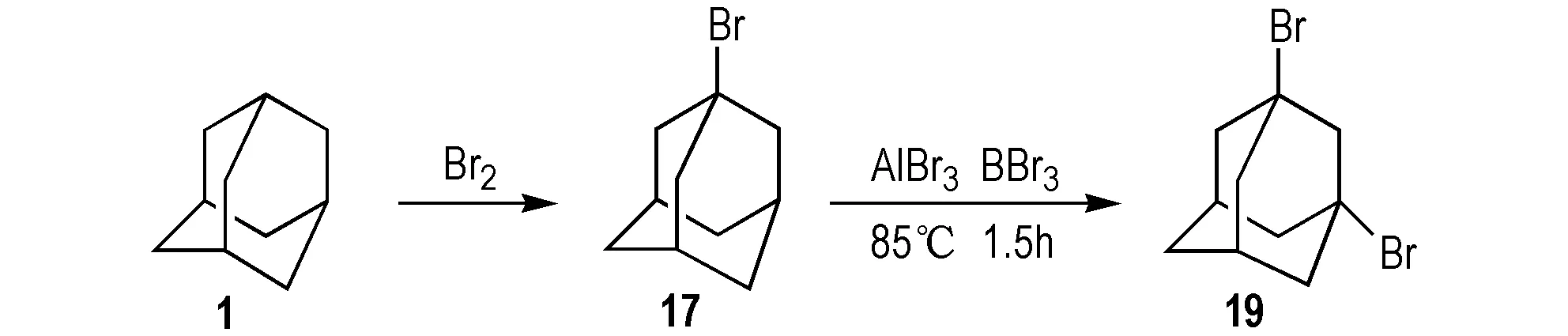
Scheme 20 Two-step synthesis of 1,3-dibromoadamantane[39]
1991年,Khusnutdinov等[40]以铑作为催化剂,和二溴甲烷在200 ℃下反应40 h,一步法合成二溴金刚烷(Scheme 21)。产物中同时存在19、1,2-二溴金刚烷(21)和1,4-二溴金刚烷(22)。
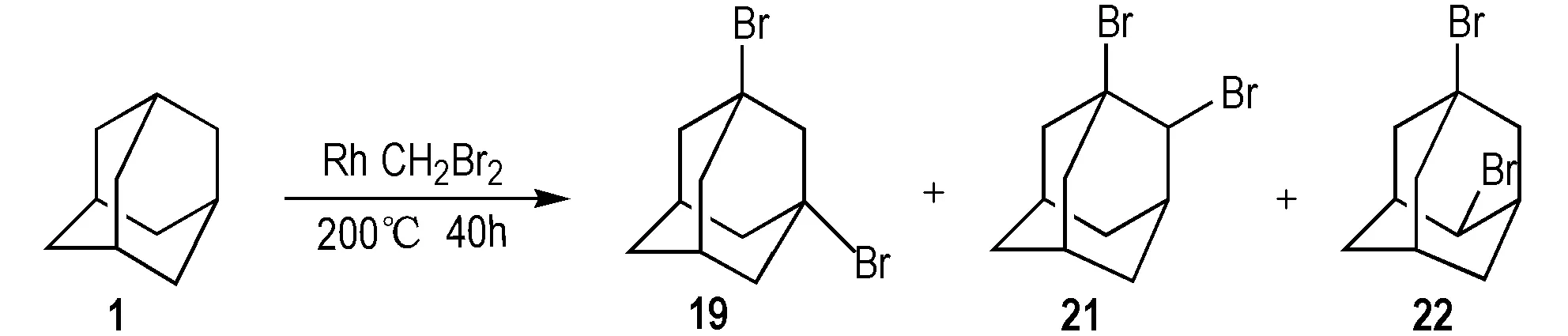
Scheme 21 One-step synthesis of dibromoadamantane[40]
2014年Degtyarenko等[41]以铁为催化剂合成19; 该法是将1与溴水在铁的催化下,20 ℃下反应2 h即可,产率高达96%; 该法催化剂廉价易得,可以重复使用,反应条件温和,将得到的19作为合成高分子材料的重要中间体。
上述路线中采用不同的催化剂合成二溴代金刚烷: 三氯化铝为催化剂两步法合成19,虽操作复杂,但采用三溴化铝催化剂,反应条件温和,产率高,反应时间短; 以铑为催化剂反应一步法合成19,催化剂价格昂贵不易得,反应温度较高,所需时间长,而且二溴甲烷在此高热下易分解为有毒的溴化氢气体,污染环境,所得产物为各种二溴代金刚烷的同分异构体,其分离提纯工艺还需进一步研究,不适合大批量生产; 以铁为催化剂价格廉价易得、选择性好、反应条件温和、时间短、产率高、因此用铁做催化剂合成19,符合现代绿色化学的需求,适合工业化生产。
2.2.3.3 1,3,5,7-四溴金刚烷
金刚烷的溴代反应选择性较高,溴代反应通常只发生在桥碳位置,1,3,5,7-四溴金刚烷(23)作为重要的有机中间体,由其可合成一系列金刚烷衍生物,而广泛的应用于医药、高分子等领域。
1980年,Sollott等[42]以1为原料与液溴在无水三氯化铝催化下反应 24 h,合成23,作为合成1,3,5,7-四硝基金刚烷的中间体。2004年,Lee等[25]同样用三氯化铝为催化剂,在Sollott的基础上将1和溴水加热反应9 h,得到23,作为合成1,3,5,7-四氨基金刚烷和1,3,5,7-四羧基金刚烷的中间体,产率为94%(Scheme 22)。2014年,Singh等[43]在Lee的基础上,将加热反应改为室温回流,得到23,为有机薄膜晶体管的中间体,产率也为94%。目前用三氯化铝作为催化剂合成23的工艺已经比较成熟。
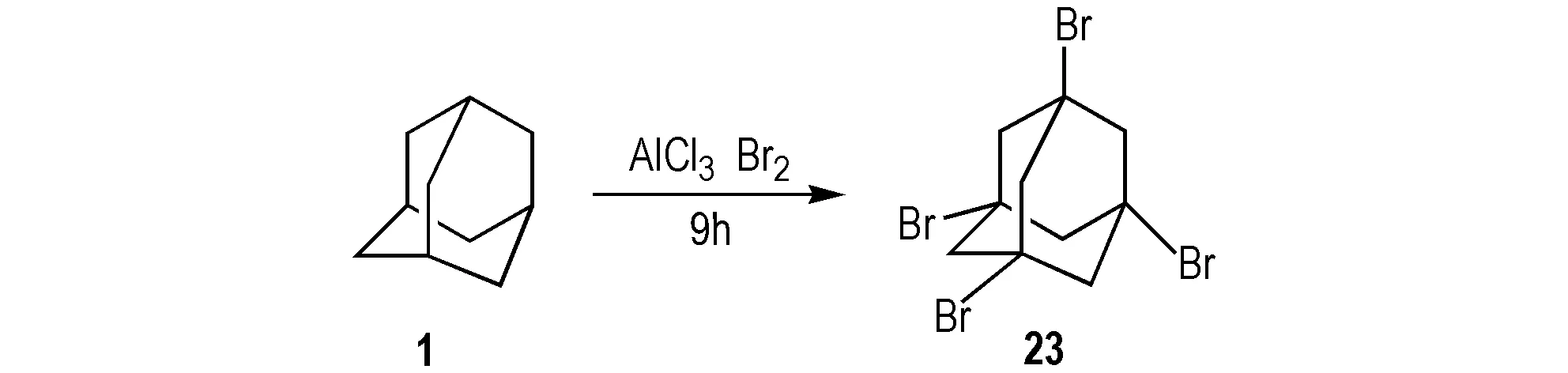
Scheme 22 Synthetic route of 1,3,5,7-tetrabrodoadamantane[25]
2.2.4 碘代金刚烷
碘代金刚烷作为重要的有机中间体,一般都是通过卤素之间的置换或者是其他物质间接反应得到,常用的碘化试剂有碘甲烷、二碘甲烷,但二碘甲烷价格昂贵,有剧毒,不适合工业化生产,尝试采用新的碘化试剂合成碘代金刚烷的反应将会是今后的研究重点。
2.2.4.1 1-碘金刚烷
1-碘金刚烷(24)可由7通过不同的碘化试剂获得。2005年,Iranpoor等[44]以7为原料,碘单质为碘化试剂,采用新的异构化试剂Silphos合成24(Scheme 23a),产率为95%。该法解决了以往由醇羟基向卤素转换过程中,五氧化二磷等副产物的分离,以及频繁的通过过柱子的方法来获得纯品的难题,其中Silphos试剂容易制备、稳定、成本低、产率高。2006年,Tajik等[45]同样以7为原料,碘化钾为碘代试剂和硫酸氢铝在己烷溶剂中,20 ℃下反应0.5 h,得到24(Scheme 23b),产率为90%。2012年Dobmeier等[46]以7为原料反应合成24(Scheme 23c),该法用氢碘酸代替原来的强酸,在水和苯两相介质中,通入惰性气体保护,80 ℃反应8 h得到产物,产率为98%。该法产率较高,但是反应时间较长,而且碘酸价格昂贵,反应过程中产生污染环境的废液,不适合工业化生产。
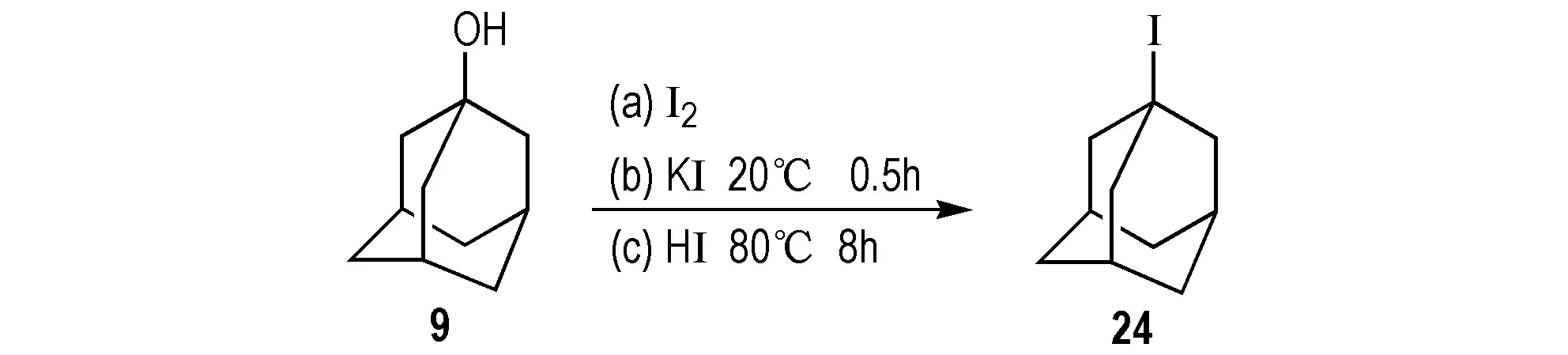
Scheme 23 Synthetic route of 1-iodoadamantane by 1-adamantanol[44-46]
Namavari等[47]以17为原料,碘酸为碘化试剂,105 ℃下反应0.5 h,合成24(Scheme 24a )。2015年Hwang等[48]以17为原料、碘为碘化试剂和四氢呋喃、氰化亚铜反应合成24(Scheme 24b),产率为85%。该法中氰化亚铜毒性较大,在空气中吸收水和二氧化碳可产生剧毒气体,危险系数较高,具有一定的局限性。
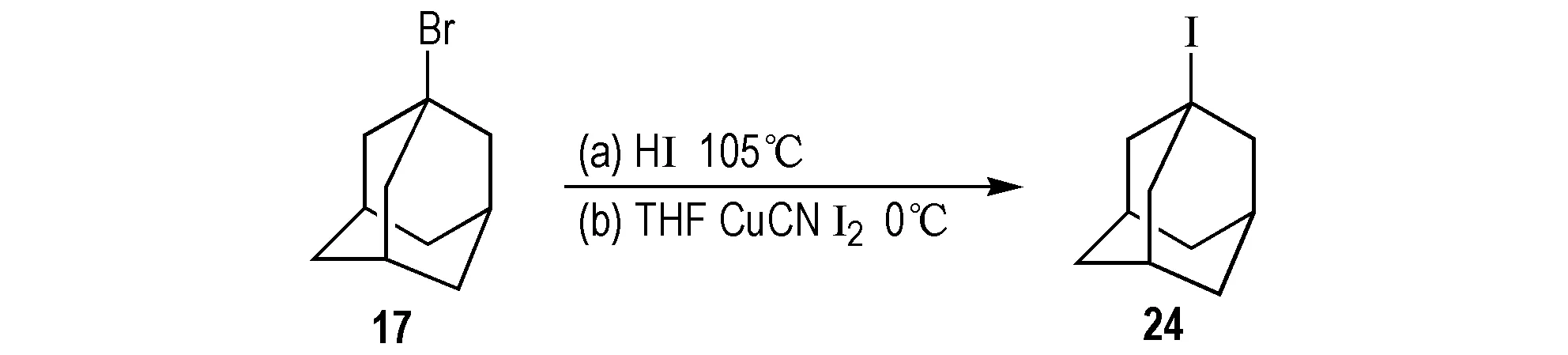
Scheme 24 Synthetic route of 1-iodoadamantane by 1-bromoadamantane[47-48]
以8为原料可以通过不同的碘化试剂反应合成24。Olah等[49]以8为原料,碘化银为碘化试剂,在氯仿溶液中加热反应16 h合成24(Scheme 25a),产率为94%,纯度大于98%。2011年Melzig等[50]以1为原料和四氢呋喃在铁为催化下反应24 h合成24(Scheme 25b),作为过渡金属催化剂的中间体,产率为61%。2013年Namavari等[47]在上述基础上以8为原料和碘酸在120 ℃的条件下反应2 h合成24(Scheme 25c),产率为92%,大大缩短了反应时间。
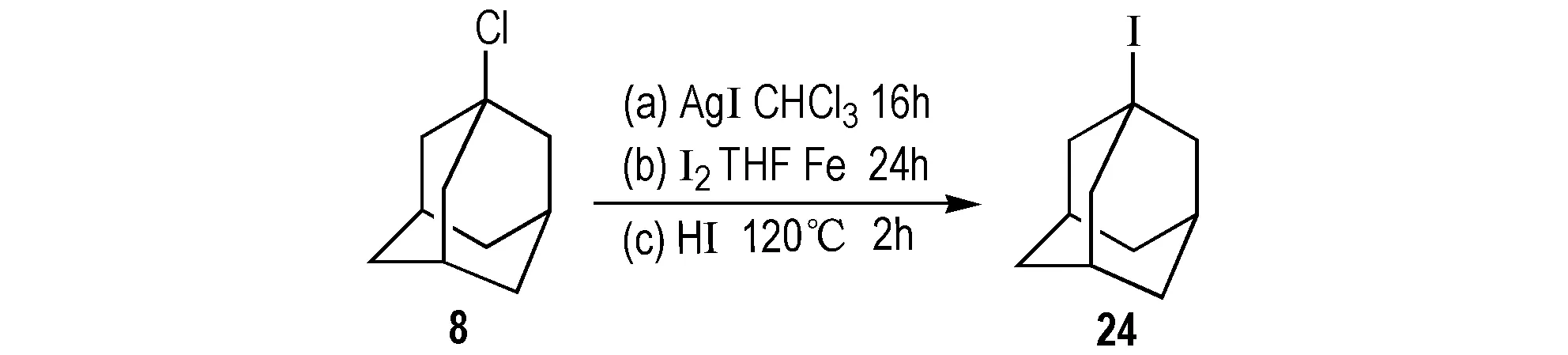
Scheme 25 Synthetic route of 1-iodoadamantane by 1-chloroadamantane[47,49,50]
2013年,Traeff等[51]以3为原料,YbI3为碘化试剂室温下反应5 min合成20(Scheme 26); 该法具有较高的选择性,而且各种官能团在溶液中有很好的兼容性,反应条件温和,时间短,但原料3价格昂贵,不易得。

Scheme 26 Synthetic route of 1-iodoadamantane by 1-fluoroadamantane[51]
2.2.4.2 1,3,5,7-四碘金刚烷
1,3,5,7-四碘金刚烷(25)作为合成1,3,5,7-四硝基金刚烷、1,3,5,7-四氨基金刚烷等衍生物的重要中间体,其合成具有重要的研究意义,25通常不能通过碘化试剂与金刚烷直接反应得到,目前相关文献报道主要是通过23与不同的碘化试剂反应制得。
Gilbert等[42]在合成1,3,5,7-四硝基金刚烷时,报道了其中间体25的合成,其合成方法是1、液溴、无水三氯化铝按一定比例反应制备23,再以二碘甲烷为碘化试剂,合成25。 Murray等[52]在合成1,3,5,7-四硝基金刚烷的同时,同样以二碘甲烷为碘化试剂两步法合成25,产率为64%。Lee等[25]以碘甲烷为碘化试剂合成25,该法第一步同样是将1、液溴、三溴化铝按一定比例反应合成23,再和碘甲烷、三溴化铝常温下反应15 h,合成25,产率为91%(Scheme 27)。
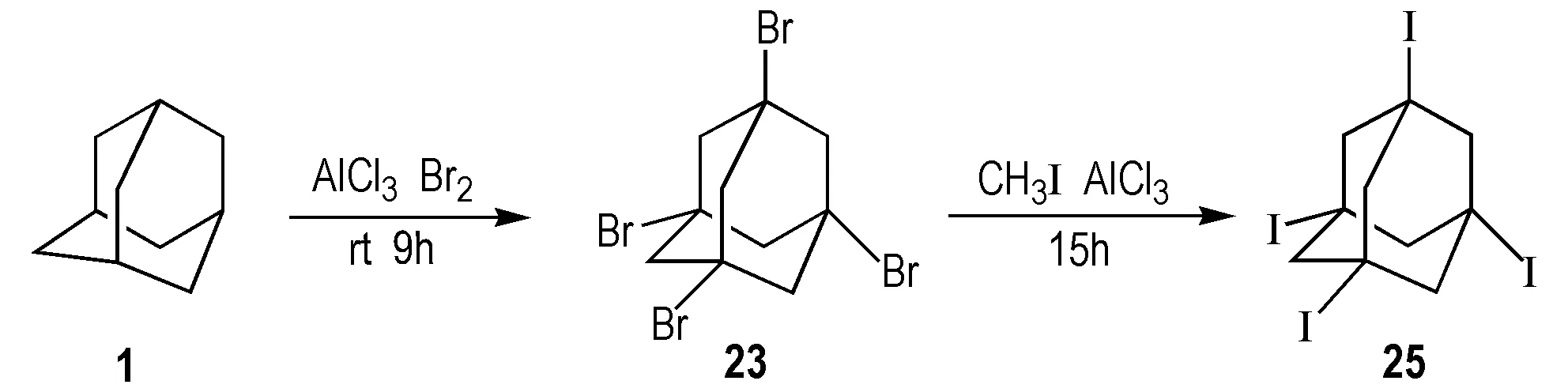
Scheme 27 Synthetic route of 1,3,5,7-tetraiodoadamantane[25]
上述路线中使用的碘甲烷、二碘甲烷等碘化试剂,实验过程中对环境有一定的危害性,不符合现代绿色化学的要求。因此,寻求新的碘化试剂和新的合成工艺路线是目前迫切需要解决的问题之一。
3 结论与展望
(1)卤代金刚烷衍生物的合成中,产物往往为各卤代物的混合物,其各卤代金刚烷的极性相差很小,寻找新的分离提纯方法,将会是今后研究的重点。
(2)金刚烷理论上最多可以引入16个卤素原子,目前国外报到最多可以引入8个卤素原子,其在国内的研究尚处于探索阶段。因此,在金刚烷分子中引入大于8个卤素原子的合成工艺路线,并探索其进一步的应用将会是今后国内外研究的热点。
(3)卤化反应中,溴水,二碘甲烷等卤化试剂,价格昂贵,有剧毒,不适合工业化生产,因此,研发一些低毒、廉价、高效又绿色环保的卤化试剂和合成路线,以适应可持续发展战略的实施和绿色化学化工的实际应用显得尤为重要。
参考文献:
[1] 杜耀. 卤代金刚烷及其硝基衍生物的合成研究[D]. 山西: 中北大学. 2014.
DU Yao. Study on the synthesis of halo and nitro derivatives of adamantine[D]. ShanXi: North University of China, Taiyuan. 2014.
[2] 杨镇, 何远航. 八硝基立方烷高温热分解分子动力学模拟[J]. 物理化学学报, 2016, 32 (4): 921-928.
YANG Zhen, HE Yuan-hang. Pyrolysis of octanitrocubane via molecular dynamics simulations[J].ActaPhysico-ChimicaSinica, 2016, 32(4): 921-928.
[3] 庞思平, 申帆帆, 吕芃浩, 等. 六硝基六氮杂异伍兹烷合成工艺研究进展[J]. 兵工学报, 2014, 35(5): 726-732.
PANG Si-ping, SHEN Fan-fan, LÜ Peng-hao, et al. Research progress in synthesis of hexanitrohexaazaisowurtzitane[J].ActaArmamentarii, 2014, 35(5): 726-732.
[4] 徐晓健, 郭建维, 朱东雨, 等. 对桥头二取代金刚烷衍生物的合成[J]. 高等学校化学学报, 2014, 35(8): 1686-1690.
XU Xiao-jian, GUO Jian-wei, ZHU Dong-yu, et al. Synthseis of symmetrical disubstituted bridgehead adamantine derivatives[J].ChemicalJournalofChineseUniversities, 2014, 35(8): 1686-1690.
[5] Schleyer P R, Olah G A. Cage hydrocarbons[M]. Wiley, 1990:24.
[6] 刘卅, 郭建维. 金刚烷的结构、溶解性及热力学性质[J]. 含能材料, 2006, 14(6): 485-490.
LIU Sa, GUO Jian-wei, Structure analysis solubility and themodynamics properties of adamantine[J].ChineseJournalofEnergeticMaterials(HannengCailiao), 2006, 14(6): 485-490.
[7] 谢炳玉, 郭建维, 刘卅, 等. 含氨基的金刚烷桥头二取代化合物合成研究[J]. 有机化学, 2011, 31(4): 486-489.
XIE Bing-yu, GUO Jian-wei, LIU Sa, et al.Synthseis of briddgeheadbis-substitued amino-adamantane compounds[J].ChineseJournalofChemistry, 2011,31(4): 486-489.
[8] Fuji M, Akita M, Tanaka T. Polycarbonate copolymer and method of producing the same: US0257362[P], 2011.
[9] 魏真, 李加荣, 张奇, 等. 金刚烷类高能量密度燃料研究进展[J]. 含能材料, 2014, 22(2): 170-176.
WEI Zhen, LI Jia-rong, ZHANG Qi, et al. Review on diamondoids as high Energetic Density fuels[J].ChineseJournalofEnergeticMaterials(HannengCailiao).2014, 22(2): 170-176.
[10] 杜耀, 王艳红, 李雅津, 等, 多硝基金刚烷的合成及其理论研究进展[J]. 化学推进剂与高分子材料, 2014, 12(2): 57-63.
DU Yao, WANG Yan-hong, LI Ya-jin, et al.Progress in synthesis and theory researchof polynitroadamantanes[J].ChemicalPropellants, 2014, 12(2): 57-63.
[11] Giamalva D H, Church D F, Pryor W A. Kinetics of ozonation.6. polycyclic aliphalic hydrocarbons[J].TheJournalofOrganicChemistry, 1988, 53(15): 3429-3432.
[12] 高治, 王艳, 苏晓明, 等. 金刚烷的化学研究[J]. 广州化学, 2004, 29 (4): 59-64.
GAO Zhi, WANG Yan, SU Xiao-ming, et al. Chemical research of adamantane[J].GuangzhouChemistry, 2004, 29(4): 59-64.
[13] Patrick T B, Johri K K, White D H, et al. Replecement of the carboxylic acid function with fluorine[J].CanadianJournalofChemistry, 1986, 64(1):138-141.
[14] YIN Feng, WANG Zhen-tao, Li Zhao-dong, et al. Silver-catalyzed decarboxylative fluorination of aliphatic carboxylic acids in aqueous solution[J].JournaloftheAmericanChemicalSociety, 2012, 134(25): 10401-10404.
[15] HUANG Xiong-yi, LIU Wei, Hooker J M, et al.Targeted fluorination with the fluoride ion by manganese-catalyzed decarboxylation[J].AngewandteChemieInternationalEdition, 2015, 54(17): 5241-5245.
[16] LIU Wei, HUANG Xiong-yi, CHENG Mu-Jeng, et al. Oxidative aliphatic C—H fluorinationwith fluoride ion catalyzed by a manganese porphyrin[J].Science, 2012, 337(6100): 1322-1325.
[17] Pitts C R, Ling B, Woltornist R, et al. Triethylborane-initiated radical chain fluorination: A synthetic method derived from mechanistic insight[J].JournalofOrganicChemistry, 2014, 79(18): 8895-8899.
[18] Aoyama M, Fukuhara T.Selective fluorination of adamantanes by an electrochemical method[J].JournalofOrganicChemistry, 2008, 73(11): 4186-4189.
[19] 易文斌, 蔡春, 钱金龙. 四氟金刚烷的合成方法: CN103193587A[P], 2013.
YI Wen-bin, CAI Chun, QIAN Jin-long. Synthetic method of tetrafluoroadamantine: CN103193587A[P], 2013.
[20] Smith G W, Williams H D. Some reactions of adamantane and adamantane derivatives[J].TheJournalofOrganicChemistry, 1961, 26(7): 2207-2212.
[21] Peter K, CHEN Ju-hua. Adamantanes and related compounds.II. chlorination of adamantane by ferric chloride and antimony pentachloride[J].TheJournalofOrganicChemistry, 1971, 36(21): 3138-3145.
[22] Draksharapu A, Codol Z, Gmez Laura, et al. Spectroscopic analyses on reaction intermediates formed during chlorination of alkanes with NaOCl catalyzed by a nickel complex[J].InorganicChemistry, 2015, 54(22): 10656-10666.
[23] WANG Zhen-tao, ZHU Lin, YIN Feng, et al. Silver-catalyzed decarboxylative chlorination of aliphatic carboxylic acids[J].JournaloftheAmericanChemicalSociety, 2012, 134(9): 4258-4263.
[24] Mondal D, Li Song-ye, Bellucci L, et al. Stereoretentive chlorination of cyclic alcohols catalyzed by titanium(Ⅳ) tetrachloride: evidence for a front side attack mechanism[J].JournalofOrganicChemistry, 2013, 78(5): 2118-2127.
[25] Lee G S, Bashara J N, Sabih G, et al. Photochemical preparation of 1,3,5,7-tetracyanoadamantane and its conversion to 1,3,5,7-tetrakis(aminomethyl)adamantine[J].OrganicLetters, 2004, 6(11): 1705-1707.
[26] Singh R, Meena J S, CHANG Yu-cheng, et al. Control of active semiconducting layer packing in organic thin film transistors through synthetic tailoring of dielectric materials[J].RSCAdvances, 2014, 4(56): 29383-29392.
[27] Khusnutdinov R I, Shchadneva N A, Baiguzina A R, et al. Chlorination of adamantane and its derivatives by carbon tetrachloride in the presence of manganese-, vanadium-, and molybdenum-containing catalysts[J].PetroleumChemistry, 2004, 44(22): 126-133.
[28] Yurchenko A G,Kulik N I,Kuchar V P, et al.On the mechanism of lquid phase halogenation of anamantane derivatives[J].TetrahedronLetters, 1986, 27(12): 1399-1402.
[29] Dart M T, Carroll W A, Florjancic A S, et al. Novel compounds as cannabinoid receptor ligands and uses thereof: Wo2007/140439 A2[P], 2007.
[30] Paulshock M. Halogenated adamantanederivatives:US3218355[P],1965.
[31] Cyril A G, Sawlewicz P. The mild bromination of adamantane and (trimethylsilyl) adamantanes[J].HelveticaChimicaActa, 1988, 71(6): 1508-1510.
[32] Nishina Y, Morita J, Ohtani B. Direct bromination of hydrocarbons catalyzed by Li2MnO3under oxygen and photo-irradiation conditions[J].RSCAdvances, 2013, 3(7): 2158-2162.
[33] Khusnutdinov R I, Shchadneva N A, Khisamova L F. Bromination of adamantane and its derivatives with tetrabromomethane catalyzed by iron compounds[J].RussianJournalofOrganicChemistry, 2015, 51(2): 184-187.
[34] Schreiner PR, Lauenstein O, KolomitsynI V, et al. Selective C—H activation of aliphatic hydro-carbons under phase-transfer conditions[J].AngewChemIntEd, 1998, 37(13-14): 1895-1897.
[35] Kruppa G, Beauchamp J. Energetics and structure of the 1-and 2- adamantyl radicals and their corresponding carbonium ions by photoelectron spectroscopy[J].JAmChemSoc, 1986, 108(9): 2162-2169.
[36] Kawakami K, Kazumitsu, Khisamova L F. Brominated methanes as photoresponsive molecular storage of elemental Br2[J].Chemistry-AnAsianJournal, 2012, 7(10): 2240-2252.
[37] Inomata S, Harada Y, Matsuoka S, et al. Synthesis and ring-opening reaction of novel 1,3-dehydroadamantanes possessing phenyl and alkoxyl substituents[J].Tetrahedron, 2013, 69(15): 3238-3248.
[38] Mizukami Y, Song Zhi-yi, Takahashi T. Halogen exchange reaction of aliphatic fluorine compounds with organic halides as halogen source[J].OrganicLetters, 2015, 17(24): 5942-5945.
[39] Denmark S E, Henke B R. Investigations on transition-state geometry in the aldol condensation[J].JournaloftheAmericanChemicalSociety, 1991, 113(6): 2177-2194.
[40] Khusnutdinov R I, Shchadneva N A,Dzhemilev U M. New efficient method for the synthesis of haloadamantanesinvoling rhodium rhodium complexes[J].IzvestiyaAkademiiNaukSSSR,SeriyaKhimicheskaya, 1991, 40(12): 2528-2529.
[41] Degtyarenko A S, Handke M, Kraemer K W, et al. Mixed-ligand hydroxocopper(Ⅱ)/pyridazine clusters embedded into 3D framework lattices[J].DaltonTransactions, 2014, 43(22): 8530-8542.
[42] Sollott G P, Gilbert E E. A facile route to 1,3,5,7-tetraaminoadamantane. Synthesis of 1,3,5,7-tetranitroadamantane[J].TheJournalofOrganicChemistry, 1980, 45(26): 5405-5408.
[43] Singh R, Meena J S, CHANG Yu-cheng. Control of active semiconducting layer packing in organic thin film transistors through synthetic tailoring of dielectric materials[J].RSCAdvances, 2014, 4(56): 29383-29392.
[44] Iranpoor N,Firouzabadi H, Jamalian A, et al.Silicaphosphine (Silphos): a filterable reagent for the conversion of alcohols and thiols to alkyl bromides and iodides[J].Tetrahedron, 2005, 61(23): 5699-5704.
[45] Tajik H, Shirini F, Zolfigol M A, et al. Convenient and efficient method for the iodination of benzylic and aliphatic alcohols by using Al(HSO4)3/KI in nonaqueous solution[J].SyntheticCommunications, 2006, 36(1): 91-95.
[46] Dobmeier M, Herrmann J M, Lenoir D, et al. Reduction of benzylic alcohols andα-hydroxycarbonyl compounds by hydriodic acid in a biphasic reaction medium[J].BeilsteinJournalofOrganicChemistry, 2012, 8(1): 330-336.
[47] Namavari M S, Phelps M E, Barrio J R. Halogen exchange reactions between alkyl halides and aqueous hydrogen halides.A new method for preparation of alkyl halides[J].TetrahedronLetters, 1990, 31(35): 4973-4976.
[48] Hwang H S, Joo S R,Kim S H.Adamantylzinc bromides: direct preparation and application to cross-coupling reaction[J].BulletinoftheKoreanChemicalSociety, 2015, 36(11): 2769-2772.
[49] Olah G A, Narang S C, Field L D. Synthetic methods and reactions. 103. preparation of alkyl iodides from alkyl fluorides and chlorides with iodotrimethylsilane or its in situ analogues[J].JournalofOrganicChemistry, 1981, 46(18): 3727-3728.
[50] Melzig L, Diene C R, Rohbogner C J, et al. Preparation of polyfunctional zinc organometallics using an Fe- or Co-catalyzed Cl/Zn-exchange[J].OrganicLetters, 2011, 13(12): 3174-3177.
[51] Traeff A M, Janjetovic M, Ta L, et al. Selective C—F bond activation: Substitution of unactivated alkyl fluorides using YbI3[J].AngewChem, 2013, 52(46): 12073-12076.
[52] Murray R W, Rajadhyaksha S N, Mohan L. Oxidation of primary amines by dimethyldioxirane[J].JournalofOrganicChemistry, 1989, 54(24): 5783-5788.
猜你喜欢
新疆有色金属(2022年2期)2022-11-22 12:38:35
云南化工(2021年11期)2022-01-12 06:06:10
——以塔里木盆地塔中地区凝析油为例
石油实验地质(2021年5期)2021-11-01 06:55:44
地球科学与环境学报(2020年2期)2020-03-26 10:01:28
石油与天然气地质(2018年4期)2018-08-01 06:36:56
中学生数理化·高二版(2016年5期)2016-05-14 13:19:33
无机盐工业(2015年2期)2015-02-17 07:05:58
中国药理学通报(2014年2期)2014-05-09 08:22:22
无机化学学报(2014年7期)2014-02-28 17:32:16
深圳大学学报(理工版)(2011年4期)2011-05-12 07:56:38
