
端环氧基聚缩水甘油醚硝酸酯的合成及固化
2017-05-07 03:37:47韩世民张得亮薛金强尚丙坤徐琰璐
含能材料 2017年1期
王 伟, 韩世民, 张得亮, 薛金强, 尚丙坤, 徐琰璐, 王 勃
(黎明化工研究设计院有限责任公司, 河南 洛阳 471000 )
1 引 言
聚缩水甘油醚硝酸酯(PGN)作为含能粘合剂,具有能量高、密度大、富氧等特点,可广泛用于大型火箭推进剂、气体发生剂、炸药等领域[1]。国外的配方研究表明[2-4],PGN有助于提高药柱的密度和比冲。陈中娥等[5]探索了PGN的性能,其摩擦感度为0,落锤撞击能I50和静电火花能E50分别为40.9 J和152.2 J,高于聚叠氮缩水甘油醚(GAP)粘合剂(分别为11.8 J和118.5 J),认为PGN是一种低感度的含能粘合剂。
通过阳离子开环聚合制得的PGN为端羟基聚合物,以端羟基聚合物为粘合剂,已有比较成熟的固化体系,通过选用异氰酸酯和调整固化参数可获得满足不同应用要求的聚氨酯弹性体,不过对水分敏感也是异氰酸酯固化体系的突出特点[6]。PGN的单体在聚合时基本为SN2开环,因此聚合物的羟基主要是活性相对较低的仲羟基,在与异氰酸酯的反应中更易受环境水分影响[7]。相关研究也表明,PGN聚氨酯网络具有较低的降解活化能,当使用脂肪族异氰酸酯为固化剂时,所形成的PGN聚氨酯室温贮存一段时间后,可发生严重降解,成为可流动的液体[8]。
为了提高PGN的固化稳定性,研究者采用了不同的改性方法[8-11],其中英国ICI炸药公司[12]通过两步反应制得了伯羟基含量达50%的四官能度PGN,提高了PGN的活性和固化稳定性,但该法操作复杂、后处理困难,并易带来安定性方面的问题。为了克服异氰酸酯固化体系存在的问题,本研究提出环氧化改性方法,在碱性条件下将PGN的端羟基官能团转化为环氧基,制得端环氧基PGN(e-PGN),并采用环氧固化剂进行固化探索,目前尚未见类似的研究报道。
2 实验部分
2.1 试剂与仪器
缩水甘油醚硝酸酯(GN),含量≥99.5%,自制; BF3·THF,自制; 1,4-丁二醇(BDO),CH2Cl2、KOH、CH3OH、Na2CO3,咪唑、邻苯二甲酸酐,分析纯,天津科密欧化学试剂有限公司;N-乙基乙二胺、异佛尔酮二胺,工业品,进口分装。
Bruker公司VERTEX70红外光谱仪,试样溶解于二甲基甲酰胺(DMF )中,KBr盐片涂膜,TGS检测器,4 cm-1分辨率,扫描16次,扫描范围400~4000 cm-1; Bruker公司AVANCE DRX500超导核磁共振波谱仪,5 mm样品管,内标物: 四甲基硅烷(TMS); TA公司Q2000差式扫描量热仪,N2流速50 mL·min-1,升温速率10 ℃·min-1,样品量10~15 mg; 东曹公司HLC8320凝胶渗透色谱仪,色谱柱TSK gel G4000+G3000+G2300,流动相THF,流速1.0 mL·min-1,柱温40 ℃,PS标样; INSTRON5969万能材料试验机,25 ℃。
羟值测定按GJB1327A-2003中4.6.1方法配制酰化剂,在(60±1) ℃酰化反应1 h,进行电位滴定; 环氧值测定按GB/T1677-2008 盐酸-丙酮法反应,进行电位滴定; 力学性能按GB/T528-2009方法进行,测试温度25 ℃,加荷速度500 mm·min-1。
2.2 实验过程
2.2.1 PGN的合成
干燥的四口瓶中加入BDO、BF3·THF和适量CH2Cl2,搅拌反应后,滴加单体GN,控制加料速度进行聚合反应。加料结束后继续反应一段时间,加入碱液中和,然后水洗至中性。甲醇洗涤有机相,真空除溶剂和水分,得淡黄色粘稠液体PGN。
IR(cm-1):3444(—OH); 2889(—CH2); 1631,1279,994,859(—ONO2); 1122(—COC—);1H NMR(Acetone)δ: 1.61(—CCH2CH2C—); 3.50~3.81(—OCH2); 3.91(—CH); 4.08(—CHOH); 4.61~4.76(—CH2ONO2);13C NMR(Acetone)δ: 26.0~26.4(—CCH2CH2C—); 67.0(—CHOH); 68.9~71.4(—OCH2—); 72.5~74.5(—CH2ONO2); 75.2~78.5(—CH); GPC: 数均分子量(Mn)为 2543,分子量分布指数(D)为1.60。
2.2.2 e-PGN的合成
三口瓶中加入PGN,以CH2Cl2溶解后加入碱溶液,搅拌反应一段时间。静置、分液,水洗有机相至中性,减压蒸馏除溶剂和水分,得淡黄色粘稠液体e-PGN。

2.2.3 e-PGN的固化
e-PGN与固化剂混合均匀,于60 ℃真空脱气后,将反应物料浇注于模具中,在设定温度下进行固化。待试样完全固化,脱模,室温静置1周,按GB/T2568-2009方法进行力学性能测试。
3 结果与讨论
3.1 环化条件对PGN端基转化的影响
PGN的环氧化反应如Scheme 1所示。

Scheme 1 Epoxidation of PGN
在碱性条件下,PGN的端羟基与相邻的硝酸酯基脱酸成环。对比PGN环化前后的IR特征吸收,发现环化后,3444 cm-1的羟基吸收消失,对应13C NMR谱图上与羟基相连的C位移(δ67.0)消失,同时出现环氧的C位移(δ43.2~43.3,50.4)和环氧的H位移(δ2.57~2.73,3.01),表明反应所得为目标产物。
影响环化反应的主要因素包括溶剂条件、环化用碱、反应温度及反应时间等。研究中以CH2Cl2为溶剂,分别考察KOH、NaOH为环化剂对e-PGN合成结果的影响,结果见表1。
表1 环化剂对e-PGN合成的影响
Table 1 Effects of cyclizing agents on the synthesis of e-PGN

PGNMn/De-PGNKOHMn/Dyield/%NaOHMn/Dyield/%2543/1.602023/1.7585.32357/1.6392.51966/1.7981.72345/1.6392.12103/1.6880.62297/1.6591.9
由表1结果可看出,以NaOH为环化剂时,e-PGN收率较高,可达92%左右; 以KOH为环化剂时,e-PGN收率较低,且e-PGN分子量较低、分子量分布较宽。试验发现,以KOH为环化剂时,反应过程物料极易乳化,产品分离困难也可能是影响收率的重要因素; 核磁分析也发现,以KOH为环化剂时,H谱显示产物较PGN中的—CH2、—CH比例提高约10%~16%,说明反应过程有明显的断链、水解等副反应发生。
采用NaOH为环化剂的样品,以H NMR谱中—CH2ONO2和—CCH2CH2C—的位移峰积分的比值计算PGN、e-PGN分子中的重复单元数(—CH2CH(CH2ONO2)O—)分别为19.92和18.18(平均值),以此计算环氧化前后聚合物的分子量分别为2460和2349,这与GPC的测试结果基本相符。说明以NaOH为环化剂条件下,聚合物的分子结构基本保持完整。因此,PGN环化制备e-PGN时选用NaOH为环化剂较为适宜。
3.2 环氧化改性对聚合物主要性能的影响
分别测试了PGN和e-PGN的主要物化特性,结果见表2。
表2 环氧化改性前后PGN和e-PGN的主要性能
Table 2 Main properities of PGN and e-PGN before and after epoxidation modification
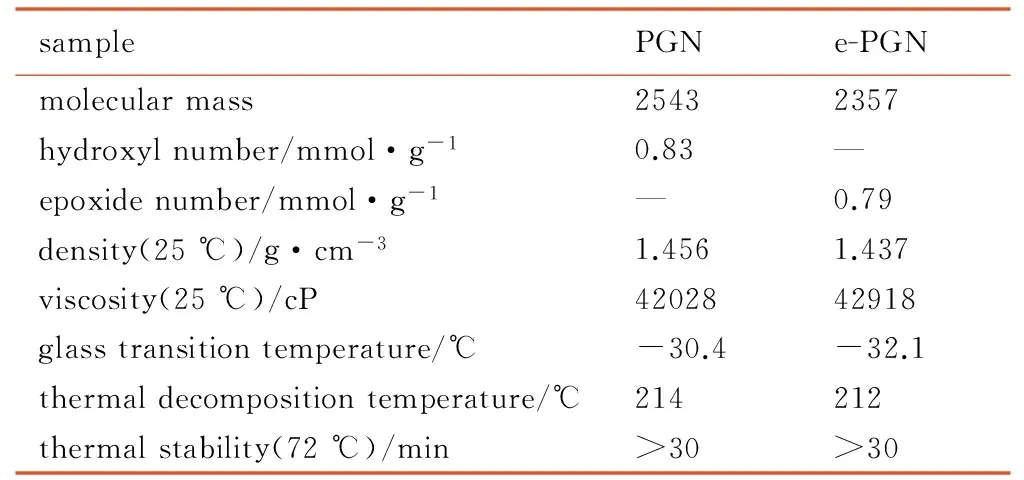
samplePGNe-PGNmolecularmass25432357hydroxylnumber/mmol·g-10.83—epoxidenumber/mmol·g-1—0.79density(25℃)/g·cm-31.4561.437viscosity(25℃)/cP4202842918glasstransitiontemperature/℃-30.4-32.1thermaldecompositiontemperature/℃214212thermalstability(72℃)/min>30>30
由表2可看出,环化前后聚合物包括粘度、玻璃化转变温度及热稳定性等在内的主要性能指标基本一致,说明端基官能团的改变,对PGN的主要性能影响不大。 e-PGN密度达到1.437 g·cm-3,基本保持了PGN的密度优势。
3.3 环氧固化剂对e-PGN固化结果的影响
分别选用不同类型的环氧固化剂进行固化试验,所选固化剂包括咪唑类的IMD、邻苯二甲酸酐(PA)及胺类的N-乙基乙二胺(NEED)、异佛尔酮二胺(IPDA)和二乙基甲苯二胺(E-100)。待物料完全固化时确定固化时间,结果见表3。
表3 固化剂对e-PGN固化反应的影响
Table 3 Effects of curing agent on e-PGN′s curing reaction
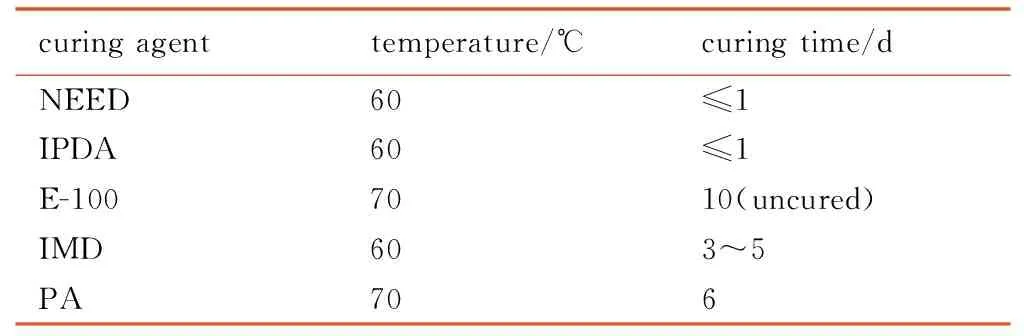
curingagenttemperature/℃curingtime/dNEED60≤1IPDA60≤1E-1007010(uncured)IMD603~5PA706
表3结果显示,固化温度为 60 ℃时,e-PGN与反应活性较高的NEED、IPDA可快速反应,混合物料在1天内即可完成固化; IMD、PA活性中等,延长固化时间和或提高固化温度至70 ℃,混合物料也可较好完成固化; 以E-100固化的试样在试验设定的温度条件下未固化,应该是固化剂活性偏低的缘故。
以e-PGN分别与IPDA、IMD、PA反应,制备环氧弹性体胶片,待胶片完全固化后,室温静置平衡7天,然后在室温和与大气接触的环境中贮存。按GB/T528-2009“硫化橡胶或热塑性橡胶拉伸应力应变性能的测定”测试胶片贮存8个月前后的力学性能,结果见表4。
表4 e-PGN胶片的力学性能
Table 4 Mechanical properties of e-PGN′s film
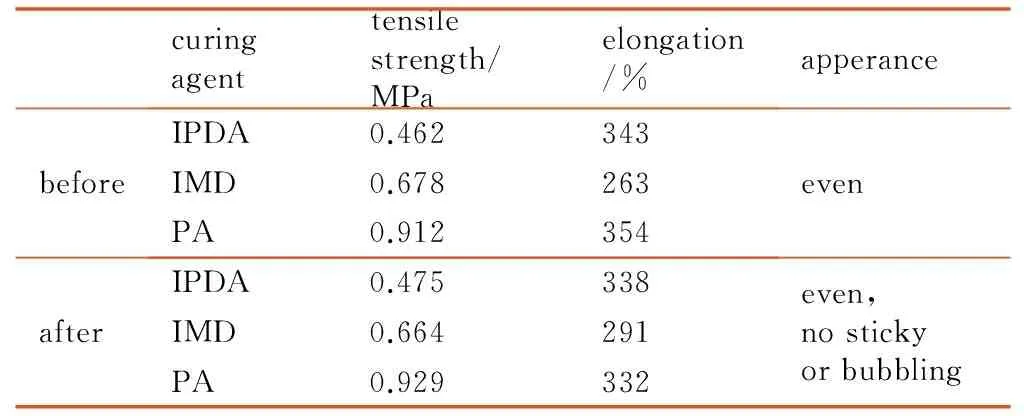
curingagenttensilestrength/MPaelongation/%apperancebeforeIPDA0.462343IMD0.678263PA0.912354evenafterIPDA0.475338IMD0.664291PA0.929332even,nostickyorbubbling
由表4可看出,e-PGN与环氧固化剂反应形成具有一定力学性能的交联网状结构,在其他条件相同的情况下,三种固化剂对应固化物的拉伸强度顺序是PA>IMD>IPDA,断裂延伸率顺序为PA>IPDA>IMD。室温贮存8个月后,三种固化胶片的力学性能基本保持不变,观察胶片与大气接触的表面亦无发粘、鼓泡等降解现象,表明e-PGN的固化物具有良好的稳定性。
4 结 论
(1) PGN经环化反应合成e-PGN,以CH2Cl2为溶剂、NaOH为环化试剂时,e-PGN收率达92%左右。
(2) PGN环氧化改性前后,包括分子量、粘度、热分解温度、玻璃化转变温度等在内的主要性能相近,e-PGN密度达1.437 g·cm-3,仍保持了PGN的密度优势。
(3) e-PGN在60 ℃/70 ℃下可与不同类型固化剂IPDA、IMD及PA等反应固化,得到具有相应力学性能的固化物; 其中以PA为固化剂时,固化物的本体力学性能最好,拉伸强度为0.912 MPa,断裂延伸率为354%。
(4) e-PGN固化胶片具有良好的稳定性,室温贮存8个月,力学性能基本稳定。
参考文献:
[1] 周劲松, 于海成, 冯渐超, 等. 聚缩水甘油醚硝酸酯合成研究进展[J]. 化学推进剂与高分子材料, 2003, 1(6): 9-12.
ZHOU Jin-song, YU Hai-cheng, FENG Jian-chao, et al. Research Progress on synthesis of the polyglycidyl ether nitrate[J].ChemicalPropellants&PolymericMaterials, 2003, 1(6): 9-12.
[2] Willer R L, McGrath D K. High performance space motor solid propellants:US5798480[P], 1998.
[3] Willer R L, McGrath D K. Clean space motor/gas generator solid propellants: US 5591936[P], 1997.
[4] Braithwaite P, Edwards W, Sanderson A, et al. The synthesis and combustion of high thermoplastic elastomer binders[C]∥32ndInternational Annual Conference of ICT, Karlsruhe, Germany, 2001: 9-1-9-7.
[5] 陈中娥, 李忠友, 姚南. PGN粘合剂的性能探索[C]∥中国航天第三专业信息网第三十三届技术交流会论文集, 张家界, 2012: 354-358.
CHENG Zhong-e, LI Zhong-you, YAO Nan. Performance researchof PGN binder[C]∥China′s Space the 33rd Technical Seminars on Professional Information Network. Zhangjiajie, 2012: 354-358.
[6] 候林法, 主编. 复合固体推进剂[M]. 北京: 宇航出版社,1994: 34.
HOU Lin-fa. Composite Solid Propellant[M]. Beijing: China Astronautic Publishing House,1994: 34。
[7] 王连心, 王伟, 崔小军, 等. 聚缩水甘油醚硝酸酯研究进展[J]. 化学推进剂与高分子材料, 2014, 12(6): 19-42.
WANG Lian-xin, WANG Wei, CUI Xiao-jun, et al. Research progress of poly(glycidyl nitrate)[J].ChemicalPropellants&PolymericMaterials, 2014, 12(6): 19-42.
[8] Bull H; Bunyan P F; Cunliffe A V, et al.An investigation into thermal stability of end-modified polyglyn[J].EnergeticMaterials-Production,ProcessingandCharacterization, 1998, 89-1.
[9] Sanderson A J, Martins L J, Dewey M A. Process for making stable cured poly (glycidyl nitrate) and energetic compositions comprising same: US6861501[P]. 2005.
[10] Kim J S, Cho J R, Lee K D, et al. 2-Nitratoethyl oxirane, poly (2-nitratoethyl oxirane) and preparation method thereof : US7880023[P]. 2011.
[11] Desai H. Studies of the cationic polymerization of vinylic and energetic cyclic ether monomers[D]. The University of Aston in Birmingham,1995.
[12] Alexander J P, Michael A D, Wayne E. One pot procedure for poly(glycidyl nitrate) end modification:US7714078B[P]. 2010.
猜你喜欢
聚氨酯工业(2022年3期)2023-01-16 13:20:54
煤矿安全(2022年9期)2022-09-16 07:25:44
吉林林业科技(2021年6期)2021-12-14 05:52:00
临床医药文献杂志(电子版)(2020年62期)2021-01-18 12:33:16
长春理工大学学报(自然科学版)(2019年6期)2020-01-07 10:43:12
国际木业(2016年9期)2017-01-15 13:55:42
中国塑料(2015年8期)2015-10-14 01:10:53
中国塑料(2015年7期)2015-10-14 01:02:38
应用化工(2015年7期)2015-04-01 01:04:08
郑州大学学报(工学版)(2015年1期)2015-03-24 01:04:43
