
O-GlcNAc糖基化修饰和Akt1对胃癌细胞体外增殖及侵袭力的影响
2017-05-03 10:08章诺贝
重庆医学 2017年8期
章诺贝,陈 新
(南昌大学第二附属医院:1.消化科;2.核医学科 330006)
论著·基础研究
O-GlcNAc糖基化修饰和Akt1对胃癌细胞体外增殖及侵袭力的影响
章诺贝1,陈 新2△
(南昌大学第二附属医院:1.消化科;2.核医学科 330006)
目的 探讨O-连接N-乙酰氨基葡萄糖(O-GlcNAc)糖基化修饰对胃癌细胞体外增殖及侵袭力的影响,并评价Akt1在O-GlcNAc糖基化促进胃癌细胞体外增殖及侵袭过程中的作用。方法 通过使O-GlcNAc转移酶(OGT)过表达(过表达OGT组)或沉默表达(沉默OGT组)及使用O-GlcNAc水解酶(OGA)特异性抑制剂Thiamet-G(抑制剂组)下调O-GlcNAc水解酶活性(抑制剂组)等构建O-GlcNAc糖基化水平升高或降低的细胞模型;采用MTT法检测各组胃癌细胞增殖活力;软琼脂集落形成实验观察各组胃癌细胞集落形成;Transwell细胞迁移实验各组胃癌细胞体外迁移和侵袭能力;蛋白免疫印迹法(Western blot)检测各组胃癌细胞Akt1活性;Thiamet-G处理Akt1表达沉默(沉默Akt1组)的胃癌细胞以评价Akt1在O-GlcNAc糖基化促进胃癌细胞侵袭中的作用;利用Thiamet-G处理Akt1过表达(过表达Akt1组)的胃癌细胞以进一步验证Akt1在O-GlcNAc糖基化调控胃癌细胞侵袭性过程中的作用。结果 O-GlcNAc糖基化水平升高可促进胃癌细胞增殖并显著提高细胞集落形成、体外迁移和侵袭的能力;Akt1活性被由O-GlcNAc糖基化水平升高所介导的Ser473磷酸化上调而激活;Thiamet-G诱导的细胞侵袭性被Akt1 shRNA所抑制;Akt1高表达可进一步促进由Thiamet-G诱导的细胞侵袭性增强。结论 O-GlcNAc糖基化可部分通过Akt1途径增强胃癌细胞的体外增殖及侵袭力。
胃肿瘤;肿瘤侵润;细胞增殖;O-GlcNAc糖基化;Akt1
O-连接N-乙酰氨基葡萄糖(O-Linked N-acetylglucosamine,O-GlcNAc)糖基化是细胞核与细胞质蛋白的丝氨酸和苏氨酸残基以O-GlcNAc修饰的一种高丰度可逆性翻译后修饰方式,它被认为能够调节细胞内蛋白质的功能及活性[1]。调控O-GlcNAc转换的酶只有两种:催化靶标蛋白分子添加GlcNAc基团的O-GlcNAc转移酶(O-GlcNAc transferase,OGT)和去除蛋白质分子中糖基的O-GlcNAc水解酶(O-GlcNAcase,OGA)。O-GlcNAc糖基化广泛参与多种细胞生物学进程,如转录、细胞生长、细胞增殖、细胞周期进程、细胞凋亡、信号转导、代谢及细胞运动等[1]。
磷脂酰肌醇3-激酶(PI3K)家族通过参与多个信号通路从而调控细胞功能。PI3K活化产生的脂质产物3,4-二磷酸磷脂酰肌醇[PI(3,4)P2]和3,4,5-三磷酸磷脂酰肌醇[PI(3,4,5)P3]作为第2信使结合并激活细胞内的靶蛋白,形成信号级联复合物,最终调控细胞的增殖、分化、生存和迁移[2]。Akt 是一种丝氨酸/苏氨酸蛋白激酶,是PI3K的下游分子。Akt至少有3个家族成员:Akt1、Akt2和Akt3,每个成员均在调节细胞功能中发挥各自的作用。PI3K/Akt信号通路在肿瘤的发生、发展中发挥重要的作用[2]。
胃癌是最常见的癌症之一,也是癌症相关性死亡的最常见原因之一。在过去的几十年中,其发病率、诊断方法和治疗手段均已发生了重要变化,但胃癌患者的预后仍然很差,尤其是在发病晚期[3]。因此,有必要进一步探讨与胃癌恶性程度相关的潜在机制。
为了确定O-GlcNAc糖基化是否参与调控胃癌的进展,笔者利用使胃癌细胞的OGT过表达、OGA抑制或OGT表达沉默等策略来改变O-GlcNAc糖基化水平,并对细胞增殖、迁移、侵袭等与恶性表型相关的细胞生物学功能进行检测。
1 材料与方法
1.1 材料
1.1.1 细胞 人胃癌细胞株AGS(ATCC CRL-1739TM,美国),人胃癌细胞株SGC-7901(凯基生物技术有限公司,江苏南京),Phoenix2TM-Ampho包装细胞(Allele生物技术公司,美国)。
1.1.2 试剂 10%胎牛血清(Wisent公司,加拿大),RPMI-1640培养基(Gibco公司,美国),FuGENE 6、蛋白酶抑制剂混合物Flag标记的人核/质OGT(ncOGT)表达载体pCMV-Flag-OGT(Sigma公司,美国),脂质体LipofectamineTM2000、遗传霉素(Invitrogen公司,美国),携带豆蔻酰化HA-标记Akt1(MAH)的pLNCX逆转录病毒载体(Addgene公司,美国),聚凝胺、Immobilon-P膜(Millipore公司,美国),PUGNAc(研究化学用品公司,加拿大),细胞裂解缓冲液(P0013,碧云天生物技术公司,江苏),O-GlcNAc单克隆抗体(RL2,亲和生物试剂公司,美国),Akt1单抗、磷酸化-Akt1单抗(细胞信号技术公司,美国),OGT多克隆抗体α、GAPDH多抗(圣克鲁斯公司,美国),RevertAidTM第一链cDNA合成试剂盒(国际Fermentas公司,立陶宛),Transwell小室(6.5 mm,Corning公司,美国),基质胶(1 mg/mL;BD生物科学公司,美国),总RNA提取试剂盒(A&A生物技术公司,波兰),电化学发光(ECL)检测试剂盒(Amersham生物科学公司,英格兰),蛋白A磁珠(通用电气医疗集团,美国)。
1.1.3 仪器 TaqMan®基因表达分析系统(应用生物系统公司,美国),Rainbow酶标仪(Tecan公司,奥地利)。
1.2 方法
1.2.1 细胞培养和处理 人胃癌细胞株AGS和SGC-7901在含有10%胎牛血清的RPMI-1640培养基中进行培养。所有细胞都在饱和湿度的37 ℃ 5%CO2培养箱中培养,细胞用5 μmol/L的Thiamet-G处理48 h或指定时间用于蛋白免疫印迹法(Western blot)或计数并传代用于侵袭性试验。
1.2.2 OGT或Akt1过表达细胞株的构建 Flag标记的人ncOGT表达载体pCMV-Flag-OGT,用于构建 OGT过表达细胞株(过表达OGT组)。使用脂质体LipofectamineTM2000按照制造商的说明书进行细胞转染。携带豆蔻酰化HA-标记Akt1(MAH)的pLNCX逆转录病毒载体用于豆蔻酰化HA-标记Akt1过表达细胞株(过表达Akt1组)的构建。Phoenix2TM-Ampho包装细胞被用作病毒载体。选用FuGENE 6作为MAH细胞的转染试剂。胃癌细胞用含10 μg/mL聚凝胺的Phoenix2TM-Ampho包装细胞的过滤培养基(病毒上清液)感染3次。选用遗传霉素作为MAH细胞的选择剂。
1.2.3 OGT或Akt1组RNA干扰细胞株的构建 将以人OGT mRNA编码序列为靶标的小干扰RNA(siRNA)用于抑制AGS和SGC-7901细胞中OGT的表达(沉默OGT组)。shOGT靶向序列是5′-GGA TGC TTA TAT CAA TTT AGG-3′。用一段shRNA寡核苷酸作为对照,靶向序列为:5′-ACG TGA CAC GTT CGG AGA ATT-3′。设计靶向人Akt1 mRNA序列编码区的siRNA并用于抑制AGS和SGC-7901细胞中Akt1的表达。靶向序列:干扰Akt1-1,5′-GCT ACT TCC TCC TCA AGA ACG-3′;干扰Akt1-2,5′-GGA CGG GCA CAT CAA GAT AAC-3′;对照为5′-ACG TGA CAC GTT CGG AGA ATT-3′。将合成的寡核苷酸退火并克隆到AgeⅠ/EcoRⅠ双酶切的pLKO.1-puro慢病毒载体。基于PLKO.1-puro载体的慢病毒通过将△8.2和VSV-G质粒共转染至HEK293T细胞包装生成。被感染的细胞用8 μg/mL的嘌呤霉素选择2周。
1.2.4 实时荧光定量PCR(RT-PCR) 用总RNA提取试剂盒按照制造商的说明书提取细胞总RNA。取1 μg细胞总RNA使用RevertAidTM第一链cDNA合成试剂盒按照试剂盒说明书逆转录获得第一链cDNAs。使用TaqMan®基因表达分析系统按照制造商的说明书进行cDNA实时扩增。采用荧光FAM标记的探针及编码Akt1和内参 GAPDH基因的序列特异性引物进行扩增。经GAPDH表达水平归一化的Akt1表达的倍数差异用公式2△△Ct进行计算。siRNA处理细胞的mRNA相对量以未经处理细胞mRNA量的百分比表示。
1.2.5 细胞增殖实验 用噻唑蓝(MTT)染料转化来评估细胞增殖。按1×104/孔将细胞接种至96孔板。让细胞生长24或48 h后,每孔加入20 μL MTT(5 mg/mL的PBS溶液)。在37 ℃经过4 h的孵育后,加入200 μL二甲基亚砜裂解细胞。使用Rainbow酶标仪测量570 nm吸光度(A)值。
1.2.6 集落形成能力试验 将细胞5×103重悬于1 mL的顶层琼脂培养基(添加0.4%琼脂的细胞培养基)。然后将细胞悬液覆盖到6孔板中1.5 mL底层琼脂培养基 (添加0.8%琼脂的细胞培养基)上,设置3个复孔。每4天向板中加入新鲜的培养基用作饲养层。2周后,在40倍光镜下选择6个视野进行集落计数。每个样本进行3次独立实验。
1.2.7 细胞迁移和侵袭试验 细胞迁移试验用带8 μm微孔膜的Transwell小室完成。将小室插入Transwell仪器。进行细胞侵袭试验时,将小室用基质胶包被。下室用600 μL含或不含OGA抑制剂的细胞条件培养液填充。将细胞5×104用100 μL含1%小牛血清的RPMI-1640培养基重悬后置于有或无OGA抑制剂的上室。20 h后,在100倍光镜下选取6个视野,对碳酸酯膜底面上出现结晶紫染色的细胞进行计数。侵袭试验中的孵化时间延长至24 h。
1.2.8 Western blot分析 用含有蛋白酶抑制剂混合物和5 μmmol/L PUGNAc(一种OGA抑制剂)的细胞裂解缓冲液裂解细胞后获取细胞总蛋白,用于Western blot分析和免疫沉淀。蛋白质样品(50 μg)用十二烷基硫酸钠-聚丙烯酰胺凝胶电泳(SDS-PAGE)在还原态下进行分离,并转移至Immobilon-P膜。检测用抗体有:O-GlcNAc单克隆抗体,Akt1单抗,磷酸化-Akt1单抗(Ser473),HA-标记单抗,OGT多克隆抗体和GAPDH多抗;用ECL检测试剂盒检测。在免疫沉淀中,细胞裂解产物与特异性抗体和蛋白A磁珠在4 ℃轻柔混合3 h。免疫沉淀物用裂解缓冲液清洗,用SDS样品缓冲液洗脱,之后用SDS-PAGE分离。检测磷酸化Akt1时,细胞裂解物中的Akt1蛋白用Akt1抗体免疫沉淀。
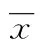
2 结 果
2.1 胃癌细胞株的O-GlcNAc糖基化水平被升高或降低 为了探讨O-GlcNAc糖基化是否对胃癌恶性程度具有重要的作用,以过表达OGT或抑制OGA(5 μmmol/L Thiamet-G处理,抑制剂组)来提高胃癌细胞株AGS和SGC-7901的O-GlcNAc糖基化水平,并通过沉默OGT表达来降低O-GlcNAc糖基化水平。(1)与未经任何处理的AGS和SGC-7901细胞(对照组)相比,过表达OGT组与经Thiamet-G处理(抑制剂组),在24 h后,O-GlcNAc糖基化水平有效升高。(2)与对照组相比,感染CGT siRNA的沉默OGT组AGS和SGC-7901细胞中OGT蛋白表达和O-GlcNAc糖基化水平均显著降低,见图1。
A:AGS细胞;B:SGC-7901。
图1 OGT过表达和OGA受抑制或沉默
OGT表达O-GlcNAc修饰水平降低
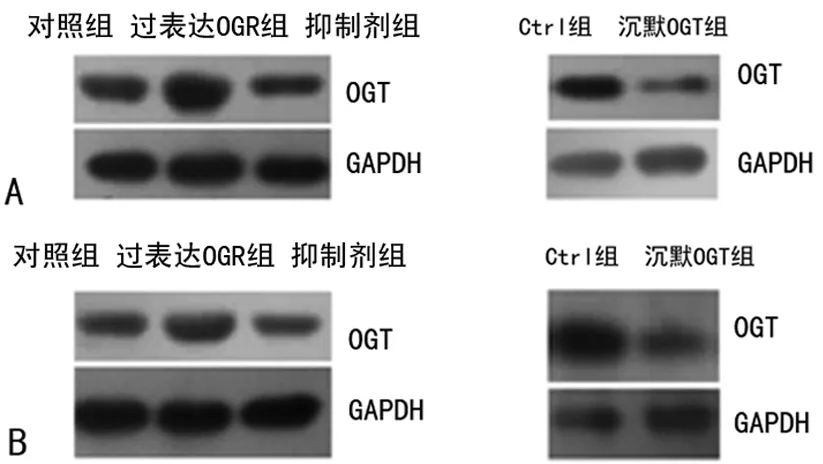
2.2 O-GlcNAc糖基化促进胃癌细胞的增殖活力 各组细胞相应处理24 h和48 h后,用MTT法检测细胞增殖率。结果表明,与对照组细胞相比,处理24 h的胃癌细胞株AGS和SGC-7901的增殖率受O-GlcNAc糖基化的影响并不明显。然而,48 h后与对照组细胞相比,过表达OGT组、抑制剂组AGS和SGC-7901细胞株增殖率分别增加约45%、22%和44%、27%。相反,沉默OGT组两种细胞的增殖率分别降低约34%和25%,见图2。
2.3 O-GlcNAc糖基化增强胃癌细胞集落形成能力 软琼脂集落试验表明,与对照组细胞相比,两种细胞中抑制剂组细胞集落形成能力增加;同时,与对照组细胞相比,沉默OGT组细胞集落形成能力降低,见图3。
A:AGS细胞;B:SGC-7901细胞;a:P<0.05;b:P<0.01,与对照组比较。
图2 O-GlcNAc修饰可促进胃癌细胞增殖
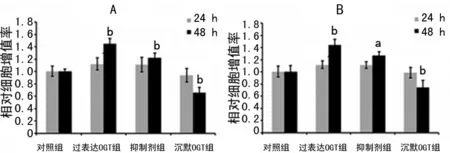
2.4 O-GlcNAc糖基化促进胃癌细胞迁移 迁移试验显示,与对照组细胞相比,过表达OGT组和抑制剂组细胞迁移能力增加,而沉默OGT组细胞的迁移能力明显抑制,见图4。
2.5 O-GlcNAc糖基化增强胃癌细胞侵袭性 O-GlcNAc糖基化在胃癌细胞转移中的作用也通过24 h体外侵袭试验进行检测。结果表明,与对照组细胞相比,过表达OGT和抑制剂组的侵袭性增强,相反,沉默OGT组细胞侵袭性明显受抑制,见图5。

a:P<0.01,与对照组比较。
图3 O-GlcNAc糖基化可增强胃癌细胞集落形成能力

a:P<0.01,与对照组比较。
图4 O-GlcNAc糖基化可促进胃癌细胞体外迁移
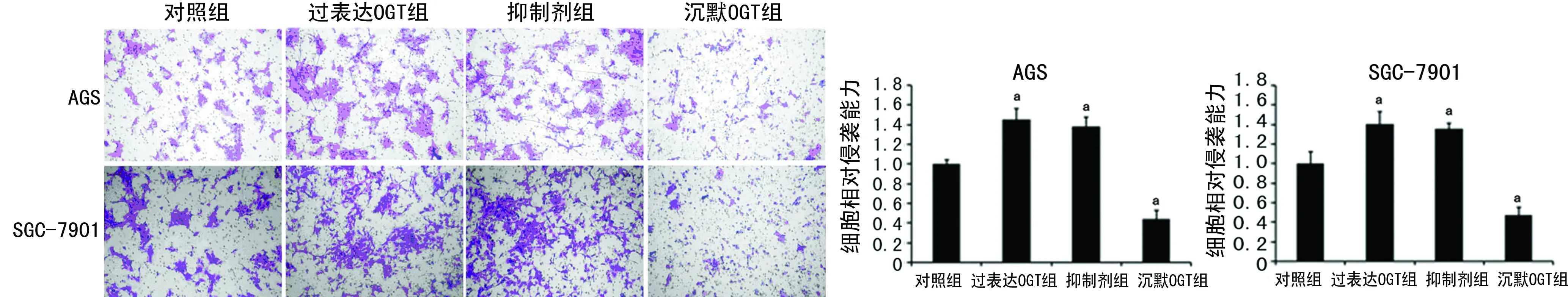
a:P<0.01,与对照组比较。
图5 O-GlcNAc糖基化可促进胃癌细胞体外侵袭性
2.6 上调O-GlcNAc糖基化会提高Akt1 Ser473位点磷酸化水平 与对照组细胞相比,抑制剂组细胞Akt1 Ser473位点磷酸化水平增加(AGS:t=2.764,P=0.031;SGC-7901:t=2.586,P=0.027),沉默OGT组细胞的Ser473位点磷酸化水平降低(AGS:t=2.694,P=0.029;SGC-7901:t=2.766,P=0.027),见图6。
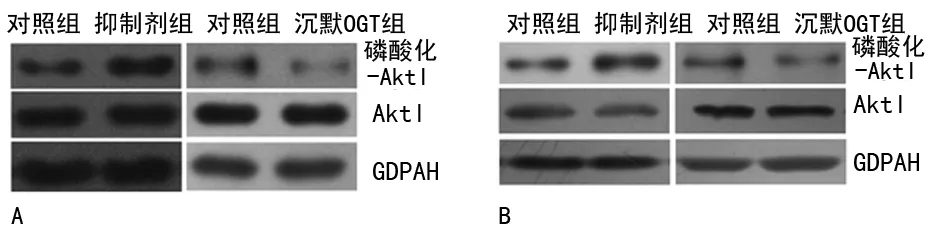
A:AGS细胞;B:SGC-7901细胞。
图6 O-GlcNAc糖基化对胃癌细胞侵袭性
的调控与PI3K/Akt1信号通路有关
2.7 胃癌细胞中Akt1蛋白的沉默 为了评价Akt1在O-GlcNAc糖基化调控胃癌细胞侵袭性中的作用,本研究用两个siRNA (干扰Akt1-1和干扰Alt1-2)下调AGS和SGC-7901细胞株中Akt1的表达水平。对照细胞则转染非沉默乱序RNA(Ctrl组)。用实时RT-PCR和Western blot分析评估RNAi的沉默效果。两个siRNA均可明显降低Akt1 mRNA水平和蛋白表达水平(图7)。然而,与siAkt1-1比较,siAkt1-2在降低Akt1水平上更为有效,因此其被选择用于后续实验(沉默Akt1组)。
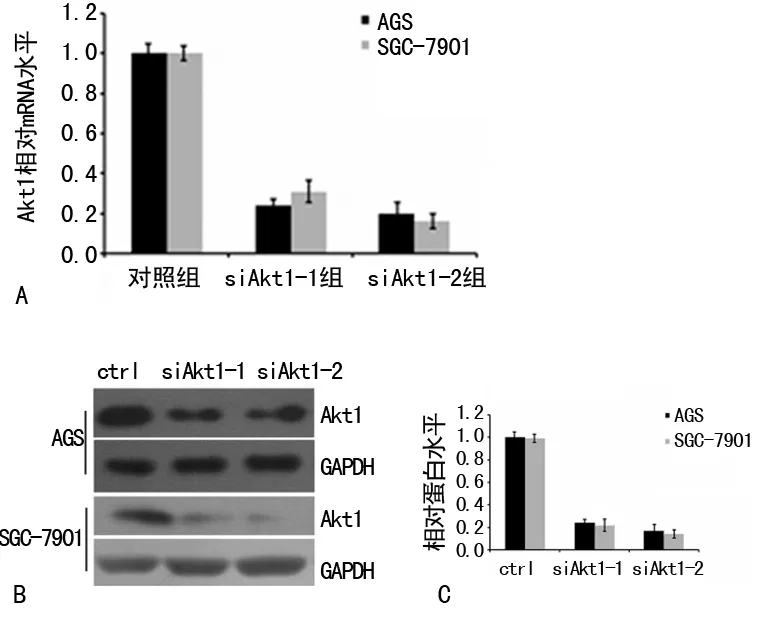
A:Akt1 mRNA分析图;B:Akt1 Western blot图。
图7 siRNA沉默胃癌细胞Akt1基因的表达
2.8 Akt1沉默可阻断O-GlcNAc糖基化对胃癌细胞侵袭性的促进作用 Akt1表达沉默的AGS细胞株用5 μmol/L Thiamet-G处理以评估Akt1对O-GlcNAc糖基化增强胃癌细胞侵袭性的影响。结果表明,Akt1表达沉默可显著减弱由Thiamet-G处理所诱导的细胞侵袭性,但其侵袭性仍然高于仅转染Akt1 shRNA的细胞。换言之,Thiamet-G能恢复Akt1表达沉默胃癌细胞的侵袭性,但仍相对低于仅被Thiamet-G处理的细胞而又相对高于对照组细胞(图8C、D)。Western blot分析(图8B)结果显示了Akt1蛋白水平及其 Ser473位点磷酸化水平。在SGC-7901细胞株也得到了类似结果(图8B)。
2.9 Akt信号通路激活可增强O-GlcNAc糖基化对胃癌细胞侵袭性作用 为进一步验证Akt1在O-GlcNAc糖基化调控胃癌细胞侵袭性过程中的作用,笔者向AGS和SGC-7901细胞株转染携带HA-标记豆蔻酰化Akt1(MAH组)的表达载体。发现相对于对照组细胞,MAH组细胞株显示出高水平的Akt1表达(图9A)。将MAH细胞用5 μmol/L Thiamet-G处理后检测其侵袭性,结果表明,Akt1过表达可进一步增强由Thiamet-G所诱导的细胞侵袭性(图9B),此结果进一步支持了上述关于Akt1与O-GlcNAc糖基化一齐促进胃癌细胞侵袭性相关的结论。

A:AGS细胞侵袭性试验分析图;B:AGS细胞Western blot;C:Western blot分析图;a:P<0.05,b:P<0.01。
图8 沉默Akt1基因表达可阻遏O-GlcNAc糖基化对胃癌细胞侵袭性的促进作用

A:Western blot;B:Western blot分析图。a:P<0.05,b:P<0.01。
图9 促进Akt1基因表达可增强O-GlcNAc糖基化对胃癌细胞侵袭性的促进作用
3 讨 论
胃癌在最常见恶性肿瘤类型中排第4位,而且是最常见的癌症死亡原因之一。尽管早期胃癌的临床治疗已经取得了重大成效,但晚期胃癌患者的长期存活率仍然相当有限。晚期或转移性胃癌的5年生存率是仅为5%~20%,中位生存期少于1年[4]。因此,调控胃癌发生、发展的确切分子机制有待进一步探索。
研究发现,O-GlcNAc糖基化在多种人类癌症的发生、发展中发挥关键作用。许多原癌基因和抑癌基因如c-Fos、c-Jun、c-Myc、pRB和p53均被 O-GlcNAc修饰[5]。Gu等[6]发现,O-GlcNAc和OGT蛋白表达水平在高转移性乳腺癌细胞系中显著升高;此外,Mi等[7]也指出,乳腺肿瘤组织中O-GlcNAc表达水平明显高于与之配对的相邻组织。以上研究结果提示,高O-GlcNAc糖基化水平可能参与乳腺肿瘤的发生发展。另外,还有报道指出,O-GlcNAc与胰腺癌、前列腺癌、卵巢癌恶性程度的调控相关[8-10]。综上所述,O-GlcNAc糖基化对肿瘤的发生发展起到了关键的调控作用。
然而,O-GlcNAc糖基化同胃癌的关系目前尚不清楚。在本研究中,笔者分析了O-GlcNAc糖基化在胃癌发生、发展中的作用。本研究结果表明,O-GlcNAc糖基化通过促进体外细胞增殖、锚定非依赖性生长、迁移和侵袭进而增强胃癌恶性程度。不过,胃癌细胞的增殖在O-GlcNAc糖基化水平升高或降低处理24 h并未受到显著影响(图2),而细胞的迁移能力和侵袭性在O-GlcNAc糖基化升高或降低处理24 h却均被促进或抑制,这提示细胞迁移能力和侵袭性受O-GlcNAc糖基化的调控可能并不依赖于其对细胞活力的影响。而在处理48 h后,O-GlcNAc糖基化则表现出对细胞增殖的调控作用。这些结果提示,O-GlcNAc糖基化水平升高可能会启动和促进胃癌的形成和转移。并且,一系列有关O-GlcNAc糖基化在肿瘤发展中作用的研究均支持了本研究结果[5-11]。
作为一种应激传感器,O-GlcNAc糖基化水平在受不同应激作用的多种哺乳动物细胞株中出现迅速且动态性升高,细胞借此重建其代谢和信号途径以促进生存[12]。肿瘤细胞的异常代谢或生长由多种形式的刺激和应激作用引起,它们包括活性氧、细胞外基质成分、基底膜、营养缺乏、缺氧和免疫系统攻击。O-GlcNAc糖基化可通过增强FOXO4转录活性从而保护细胞免受氧化应激损伤[13]。此外,通过降低OGT表达水平或阻断己糖胺生物合成途径(hexosamine biosynthetic pathway,HBP)来降低 O-GlcNAc糖基化能够使细胞对凋亡刺激更加敏感[12]。因此,O-GlcNAc糖基化水平升高也可能有利于胃癌细胞抵抗凋亡刺激,从而加速肿瘤的形成和发展。
PI3K/Akt信号通路可通过多种途径传递侵袭和转移信号并且还参与多种因素介导的肿瘤血管生成过程。PI3K通过与E-钙黏蛋白、β-连环蛋白及血管内皮生长因子受体2(VEGFR-2)相互作用从而参与血管内皮生长因子(VEGF)介导的内皮细胞信号,进而激活PI3K/Akt通路;此外,VEGFR-2和αVβ3复合体也可通过PI3K依赖信号通路介导内皮细胞的黏附和迁移[14];PI3K/Akt还能够通过促进由肿瘤坏死因子诱导的内皮细胞迁移从而调节肿瘤血管生成[15]。有报道指出,O-GlcNAc糖基化的上调会通过增加Akt1活性从而促进甲状腺未分化癌细胞的增殖[16]。本研究发现O-GlcNAc糖基化能提高胃癌细胞的侵袭性。Akt1表达沉默可减弱由Thiamet-G所诱导的细胞侵袭性,但其侵袭性仍然仅高于Akt1表达沉默的细胞(图8A、C)。此外,Akt1过表达还能够增强由Thiamet-G所诱导的细胞侵袭性。以上结果提示,O-GlcNAc糖基化可部分经Akt1调节胃癌细胞侵袭性,不过,还可能有其他信号通路或蛋白质参与该调控过程。细胞表面E-钙黏蛋白的减少和细胞黏附的减弱可能是 O-GlcNAc糖基化诱导乳腺癌转移的潜在分子机制[7]。肌动蛋白结合蛋白-人肌动蛋白素(cofilin)在OGT的作用下发生O-GlcNAc糖基化,并在由OGT调控的细胞迁移过程中发挥关键的介导作用[17]。在乳腺癌细胞三维细胞侵袭过程中,cofilin的Ser-108位点的O-GlcNAc糖基化是其正确定位于乳腺癌细胞前缘侵袭性伪足的必要条件[17]。近年来,越来越多的证据表明,O-GlcNAc糖基化的靶标蛋白和O-GlcNAc调节的信号通路与肿瘤发生、发展有关[8-11]。因此,有必要进一步深入研究以更好地理解由 O-GlcNAc糖基化诱导的胃癌侵袭的潜在机制。此外,Akt至少包括3个家族成员:Akt1、Akt2和Akt3,每个成员在调节细胞功能中均发挥各自作用。除Akt1外,其他Akt家族成员的活性变化对O-GlcNAc糖基化调控细胞恶性的影响仍有待阐明。
总之,本研究提供的以上证据表明,O-GlcNAc糖基化能够影响胃癌的恶性程度。显然,理解异常O-GlcNAc糖基化与肿瘤生物学行为之间的明确关系必将有利于癌症的诊断和治疗。
[1]Hart GW,Housley MP,Slawson C.Cycling of O-linked beta-N-acetylglucosamine on nucleocytoplasmic proteins[J].Nature,2007,446(7139):1017-1022.
[2]Arboleda MJ,Lyons JF,Kabbinavar FF,et al.Overexpression of AKT2/protein kinase B beta leads to up-regulation of beta 1 integrins,increased invasion,and metastasis of human breast and ovarian cancer cells[J].Cancer Res,2003,63(1):196-206.
[3]Herrero R,Park JY,Forman D.The fight against gastric cancer-the IARC Working Group report[J].Best Pract Res Clin Gastroenterol,2014,28(6):1107-1114.
[4]Kamangar F,Dores GM,Anderson WF.Patterns of cancer incidence,mortality,and prevalence across five continents:defining priorities to reduce cancer disparities in different geographic regions of the world[J].J Clin Oncol,2006,24(14):2137-2150.
[5]Kamemura K,Hayes BK,Comer FI,et al.Dynamic interplay between O-glycosylation and O-phosphorylation of nucleocytoplasmic proteins:alternative glycosylation/phosphorylation of THR-58,a known mutational hot spot of c-Myc in lymphomas,is regulated by mitogens[J].J Biol Chem,2002,277(21):19229-19235.
[6]Gu Y,Mi W,Ge Y,et al.GlcNAcylation plays an essential role in breast cancer metastasis[J].Cancer Res,2010,70(15):6344-6351.
[7]Mi W,Gu Y,Han C,et al.O-GlcNAcylation is a novel regulator of lung and colon cancer malignancy[J].Biochim Biophys Acta,2011,1812(4):514-519.
[8]Banerjee S,Sangwan V,McGinn O,et al.Triptolide-induced cell death in pancreatic cancer is mediated by O-GlcNAc modification of transcription factor Sp1[J].J Biol Chem,2013,288(47):33927-33938.
[9]Kamigaito T,Okaneya T,Kawakubo M,et al.Overexpression of O-GlcNAc by prostate cancer cells is significantly associated with poor prognosis of patients[J].Prostate Cancer Prostatic Dis,2014,17(1):18-22.
[10]de Queiroz RM,Madan R,Chien J,et al.Changes in O-linked N-acetylglucosamine (O-GlcNAc) homeostasis activate the p53 pathway in ovarian cancer cells[J].J Biol Chem,2016,291(36):18897-18914.
[11]Ma Z,Vosseller K.Cancer metabolism and elevated O-GlcNAc in oncogenic signaling[J].J Biol Chem,2014,289(50):34457-34465.
[12]Sohn KC,Lee KY,Park JE,et al.OGT functions as a catalytic chaperone under heat stress response:a unique defense role of OGT in hyperthermia[J].Biochem Biophys Res Commun,2004,322(3):1045-1051.
[13]HoSR,WangK,WhisenhuntTR,etal.O-GlcNAcylationenhancesFOXO4transcriptionalregu-lation in response to stress[J].FEBS Lett,2010,584(1):49-54.
[14]Tan PH,Xue SA,Manunta M,et al.Effect of vectors on human endothelial cell signal transduction:implications for cardiovascular gene therapy[J].Arterioscler Thromb Vasc Biol,2006,26(3):462-467.
[15]Zhang R,Xu Y,Ekman N,et al.Etk/Bmx transactivates vascular endothelial growth factor 2 and recruits phosphatidylinositol 3-kinase to mediate the tumor necrosis factor-induced angiogenic pathway[J].J Biol Chem,2003,278(51):51267-51276.
[16]Krzeslak A,Jóžwiak P,Lipińska A.Down-regulation of β-N-acetyl-D-glucosaminidase increases Akt1 activity in thyroid anaplastic cancer cells[J].Oncol Rep,2011,26(3):743-749.
[17]Huang X,Pan Q,Sun D,et al.O-GlcNAcylation of cofilin promotes breast cancer cell invasion[J].J Biol Chem,2013,288(51):36418-36425.
Effects of O-GlcNAcylation modification and Akt1 on proliferation and invasion of gastric cancer cells
ZhangNuobei1,ChenXin2△
(1.DepartmentofGastroenterology;2.DepartmentofNuclearMedicine,theSecondAffiliatedHospitalofNanchangUniversity,Nanchang,Jiangxi330006,China)
Objective To study the influence of O-GlcNAcylation on on proliferation and invasion of gastric cancer cells and evaluate the role of Akt1 on O-GlcNAcylation promotting cells proliferation and invasion in gastric cancer.Methods Build the cell model:O-GlcNAc glycosylation levels rise or fall.The cell viability was determine by MTT.To investigate whether O-GlcNAcylation affected colony formation ability of gastric cancer cells,soft agar colony assays were carried out.Cell migration or invasion was using transwell chambers.The expression of Akt1 was detected through Western blot.Thiamet-G was used to eualuate the role of Akt1 on O-Gcnac cylation regulating invasion in gastric Cancei.Results O-GlcNAcylation was increased the gastric cancer cells proliferation ability,colony formation ability,migration and invasion ability in vitro.Akt1 was activated by Ser473 phosphorylation upregulation though O-GlcNAcylation.Akt1 shRNA was inhibition the cell invasive which induced by Thiamet-G.Akt1 overexpression was promoted by Thiamet-G-induced cell invasion.Conclusion O-GlcNAcylation enhanced oncogenic phenotypes possibly partially involving Akt1.
stomach neoplasms;neoplasm invasiveness;cell proliferation;O-GlcNAcylation;Akt1
章诺贝(1982-),副主任医师,硕士,主要从事消化系肿瘤基因治疗研究。△
,E-mail:13870979404@163.com。
10.3969/j.issn.1671-8348.2017.08.007
R735.7
A
1671-8348(2017)08-1027-05
2016-10-22
2016-12-20)
猜你喜欢
现代畜牧科技(2021年6期)2021-07-16
天然产物研究与开发(2018年7期)2018-08-21
上海农业学报(2017年3期)2017-04-10
医学研究杂志(2015年12期)2015-06-10
医学研究杂志(2015年11期)2015-06-10
中国当代医药(2015年16期)2015-03-01
中国当代医药(2015年16期)2015-03-01
中国医药导报(2015年27期)2015-02-28
中国药理学通报(2014年2期)2014-05-09
癌变·畸变·突变(2014年2期)2014-03-01
