
RP-HPLC法测定复方奥美拉唑干混悬剂的含量及有关物质
2017-04-26 11:55田晓峰王国军
实用药物与临床 2017年4期
田晓峰,王国军
RP-HPLC法测定复方奥美拉唑干混悬剂的含量及有关物质
田晓峰*,王国军
目的 建立RP-HPLC法测定复方奥美拉唑干混悬剂中奥美拉唑的含量及其有关物质。方法 采用Phenomenex Luna C18色谱柱(250 mm×4.60 mm,5 μm),流动相:甲醇-水-三乙胺-磷酸(体积比为55∶45∶0.3∶0.12),流速:1.0 mL/min,柱温:30 ℃,进样量:20 μL,含量测定的检测波长为302 nm,有关物质测定的检测波长为280 nm。结果 奥美拉唑经强制破坏主峰与杂质可得到良好的分离,浓度在0.133~30.0 mg/L范围内线性关系良好(r=0.999 7),平均回收率为99.0%(RSD=0.2%,n=9)。3批样品及对照药品的含量分别为97.5%、97.6%、97.5%、92.3%,单一最大杂质分别为0.06%、0.06%、0.05%、0.15%,总杂质分别为0.11%、0.11%、0.11%、0.45%。结论 本法可用于测定复方奥美拉唑干混悬剂的含量及有关物质。
奥美拉唑;高效液相色谱法;含量测定;有关物质
0 引言
奥美拉唑(Omeprazole)是首个上市的质子泵抑制剂,由瑞典阿斯特拉制药公司(阿斯利康制药前身)研发,于1988年在瑞士上市,用于治疗十二指肠溃疡、胃溃疡、反流性食管炎及Zollinger-Ellison综合征[1-3]。复方奥美拉唑干混悬剂商品名为ZEGERID,由桑塔鲁斯(Santarus)公司研制,于2004年6月15日获批在美国上市,目前我国还没有该药上市。本品为其仿制药,由奥美拉唑和碳酸氢钠所组成,方中碳酸氢钠不仅具有抑制胃酸分泌的直接效果,还能防止奥美拉唑被胃酸降解,组方合理。而且该药服用后,药物吸收快速,可以很快达到峰浓度水平,能有效控制胃酸。
为了确保本品的质量,笔者依据《中国药典》2010版二部、《英国药典》(BP2009)中奥美拉唑质量标准及相关文献[4-9],将奥美拉唑质量标准中含量及有关物质测定的色谱条件进行优化,并建立本品奥美拉唑含量及有关物质测定的方法,为复方奥美拉唑干混悬剂及其相关制剂的质量控制提供科学依据。
1 仪器与试药
美国Agilent 1100型高效液相色谱仪(DAD检测器,Chemstation system化学工作站),瑞士Mettler AE240十万分之一电子天平。
奥美拉唑对照品(中国食品药品检定研究所,批号:100367-201104),复方奥美拉唑干混悬剂(海南天煌制药有限公司,规格:奥美拉唑20 mg,批号:20111101、20111102、20111103),对照药品:ZEGERID(Santarus公司,规格:奥美拉唑20 mg,批号:C1H45801),奥美拉唑磺酰化物对照品(杂质D,中国食品药品检定研究所,批号:100838-201103),杂质A、B、E、H、I(均由深圳菲斯化工有限公司提供,批号分别为2011030211、20110321、20110407、1458-030A6、20110402),娃哈哈纯净水(杭州娃哈哈集团有限公司),甲醇、乙腈(天津康科德科技有限公司,色谱纯),三乙胺、磷酸(天津市科密欧化学试剂有限公司,色谱纯)。
2 方法与结果
2.1 色谱条件 色谱柱:Phenomenex Luna C18柱(250 mm×4.60 mm,5 μm),流动相:甲醇-水-三乙胺-磷酸(体积比为55∶45∶0.3∶0.12),流速:1.0 mL/min,柱温:30 ℃,进样量:20 μL,含量测定的检测波长:302 nm,有关物质测定的检测波长:280 nm。
2.2 溶液的制备 对照品溶液:取奥美拉唑对照品约20 mg,精密称定,置100 mL量瓶中,加流动相溶解并稀释至刻度,摇匀。取1.0 mL置10 mL量瓶中,加流动相稀释至刻度,摇匀,滤过,制成每1 mL中约含奥美拉唑20 μg的对照品溶液。
含量测定供试品溶液:取复方奥美拉唑干混悬剂约5.85 g(约相当于奥美拉唑20 mg),精密称定,置100 mL量瓶中,加流动相超声溶解并稀释至刻度,摇匀。取1.0 mL置10 mL量瓶中,加流动相稀释至刻度,摇匀,滤过,制成每1 mL中约含奥美拉唑20 μg的供试品溶液。
自身对照溶液:取奥美拉唑对照品约20 mg,精密称定,置100 mL量瓶中,加流动相溶解并稀释至刻度,摇匀。取1.0 mL置100 mL量瓶中,加流动相稀释至刻度,摇匀,滤过,制成每1 mL中约含奥美拉唑2 μg的对照溶液。
有关物质测定供试品溶液:取复方奥美拉唑干混悬剂约5.85 g(约相当于奥美拉唑20 mg),精密称定,置100 mL量瓶中,加流动相超声溶解并稀释至刻度,摇匀,滤过,制成每1 mL中约含奥美拉唑200 μg的供试品溶液。
2.3 系统适用性 取奥美拉唑对照品及奥美拉唑磺酰化物对照品,加流动相稀释制成每1 mL中约含奥美拉唑及奥美拉唑磺酰化物各100 μg的混合对照品溶液,取20 μL注入液相色谱仪,奥美拉唑峰与奥美拉唑磺酰化物峰(相对保留时间约为0.8)的分离度应>2.0(测得结果为5.7),理论板数按奥美拉唑峰计算应不低于2 000(测得结果为14 533),见图1。

图1 系统适用性溶液色谱图 注:1.奥美拉唑磺酰化物,2.奥美拉唑
2.4 专属性 取本品适量,精密称定,加流动相稀释制成约含奥美拉唑400 mg/L的储备液,对其进行强制破坏试验。①酸破坏:精密量取储备液5 mL,置10 mL量瓶中,加1 mol/L盐酸溶液1 mL,室温放置5 min,用1 mol/L氢氧化钠溶液调节pH值至中性,加流动相定容。②碱破坏:精密量取储备液5 mL,置10 mL量瓶中,加1 mol/L氢氧化钠溶液1 mL,室温放置6 h,加1 mol/L盐酸溶液调节pH值至中性,加流动相定容。③氧化破坏:精密量取储备液5 mL,置10 mL量瓶中,加体积分数为10%的过氧化氢溶液1 mL,室温放置 30 min,加流动相定容。④高温破坏:精密量取储备液5 mL,置10 mL量瓶中,水浴加热2 h,冷却至室温,加流动相定容。⑤光照破坏:精密量取储备液5 mL,置10 mL量瓶中,置4 500 Lx光线下放置6 h,加流动相定容。取上述溶液各20 μL进样,见图2。试验结果表明,本品主成分峰与各分解产物峰均可实现基线分离,并且同法做空白辅料的破坏试验,空白辅料对主峰含量检测无干扰,故该方法专属性良好。
2.5 溶液稳定性
2.5.1 含量测定供试品溶液稳定性 取本品样品(批号:20111101)约5.85 g,按照“2.2”项下制备含量测定供试品溶液,于室温下放置8 h,分别于0、1、2、4、8 h,取20 μL注入液相色谱仪,记录色谱图,考察样品的稳定性,结果奥美拉唑峰面积的RSD为0.2%,表明室温下放置8 h内含量测定供试品溶液稳定,满足样品含量测定要求。
2.5.2 有关物质测定供试品溶液稳定性 取本品样品(批号:20111101)约5.85 g,按照“2.2”项下制备有关物质测定供试品溶液,于室温下放置8 h,分别于0、1、2、4、8 h,取20 μL注入液相色谱仪,记录色谱图,考察样品的稳定性,结果杂质总峰面积不断增大,并且杂质数不断增多,表明本品有关物质测定供试品溶液室温下放置易降解生成新的杂质,样品需要新鲜配制检测。

图2 破坏试验色谱图 注:A.酸破坏,B.碱破坏,C.氧化破坏,D.高温破坏,E.光照破坏,F.空白
2.6 检测限和定量限 以信噪比S/N≥3时的浓度为检测限,以信噪比S/N≥10时的浓度为定量限。取对照品溶液稀释若干倍后进样20 μL,测得奥美拉唑的检测限为0.040 mg/L,定量限为0.133 mg/L。
2.7 线性范围 精密称取奥美拉唑对照品20 mg,置100 mL量瓶中,加流动相稀释至刻度,制成每1 mL含200 μg的对照品储备溶液,精密量取0.2、0.4、0.8、1.0、1.2、1.5 mL,分别置于10 mL量瓶内,加流动相溶解并稀释至刻度,得4.0、8.0、16.0、20.0、24.0、30.0 mg/L的系列对照品溶液,精密量取上述溶液及定量限溶液(0.133 mg/L)各20 μL进样,记录主成分峰面积,并以质量浓度C(mg/L)为横坐标、峰面积A为纵坐标绘制标准曲线,得回归方程为A=43.248 C-2.879 9 (r=0.999 7),结果表明,其在质量浓度0.133~30.0 mg/L范围内与峰面积线性关系良好。
2.8 精密度、重复性、准确度试验
2.8.1 仪器精密度 按“2.2”项下制备对照品溶液和自身对照溶液,分别按照“2.1”项下色谱条件连续进样6次,记录色谱图,量取峰面积,计算峰面积的RSD分别为0.1%和0.7%。在可接受范围内,表明仪器精密度良好。
2.8.2 重复性 取本品样品(批号:20111101)6份,分别按照“2.2”项下平行制备含量测定供试品溶液,按“2.1”项下色谱条件进样测定,得奥美拉唑含量的RSD为0.1%,在可接受范围内,表明方法重复性良好。
2.8.3 准确度 以回收率试验来验证准确度,按处方比例称取各辅料9份,置于100 mL量瓶中,按复方奥美拉唑干混悬剂20 mg规格标示量的80%、100%、120%,分别精密量取奥美拉唑对照品约16、20、24 mg各3份,置于上述100 mL量瓶内,加流动相溶解并稀释制成每1 mL中约含16、20、24 μg的溶液,摇匀,滤过,作为供试品溶液。精密量取上述溶液各20 μL,注入液相色谱仪,记录色谱图,量取峰面积并计算回收率,见表1。

表1 奥美拉唑回收率结果
2.9 奥美拉唑含量及有关物质测定 取3批奥美拉唑干混悬剂,按照“2.2”项下制备对照品溶液、含量测定供试品溶液、自身对照溶液及有关物质测定供试品溶液,按“2.1”项下色谱条件进样分析,记录色谱图,以外标法计算奥美拉唑的含量,以自身对照法计算有关物质的含量,结果见表2。

表2 含量及有关物质测定结果
3 讨论
3.1 色谱柱类型改变 色谱柱由以辛烷基硅烷键合硅胶的填料类型更换成以十八烷基硅烷键合硅胶的填料类型,是由于考虑到C18柱比C8柱的碳链更长,保留特性更好,并且C18柱和C8柱相比,耐用性能较好,使用寿命长,成本低。
3.2 流动相改变 流动相由乙腈-水的缓冲盐系统改为甲醇-水的缓冲盐系统,虽然有机溶剂类型有所改变,但缓冲盐的pH值范围基本保持不变,并且对样品的色谱行为影响几乎不变。
3.3 检测波长 通过全波长扫描可知,奥美拉唑与奥美拉唑杂质A、B、D、E、H、I的最大吸收峰为202、276、302 mn,由于202 nm是靠近末端吸收,不建议采用该波长。故检测波长的选择依据《中国药典》2010版二部中奥美拉唑肠溶片及奥美拉唑肠溶胶囊的含量和有关物质测定波长,分别选择302、280 nm作为含量及有关物质的检测波长。
3.4 两种方法下样品与对照药品对比 采用《中国药典》2010版二部中奥美拉唑原料药有关物质检测方法和本品有关物质检测方法,分别对自制样品与对照药品ZEGERID进行检测,见表3。
由以上对比结果可见,本品有关物质检测方法与《中国药典》2010版二部奥美拉唑原料药有关物质检测方法相比:①将保留时间提前,缩短了检测时间;②奥美拉唑与奥美拉唑磺酰化物分离度由原来的2.8提高到6.2;③检测出的杂质个数相同;④两种方法下测得的总杂质含量接近;⑤空白辅料对样品检测均不产生干扰。优化的有关物质检测方法更适用于本品有关物质检测,与《中国药典》2010版二部奥美拉唑的有关物质检测方法相比,同样能够达到检测目的。3.5 已知杂质研究 通过将《英国药典》(BP2009)有关物质中的已知杂质(奥美拉唑杂质A、B、D、E、H、I)对照品与本品原料药及制剂的杂质进行对比,确定本品中杂质是否为已知杂质。由于本品有关物质检测方法与《英国药典》(BP2009)中方法存在差异,因此采用两种方法进行对比研究,见表4。
由以上对比结果可见:①原料药中含有3个已知杂质;②制剂中含有2个已知杂质;③《英国药典》(BP2009)有关物质检测方法中杂质B和杂质D相对保留时间相同,杂质峰出现重叠现象,该方法不能将所有杂质在同一条件下较好地分离,而本品的有关物质方法可以将所有杂质在同一条件下较好地分离,更适合本品有关物质的检测。

表3 两种方法下样品与对照药品对比结果
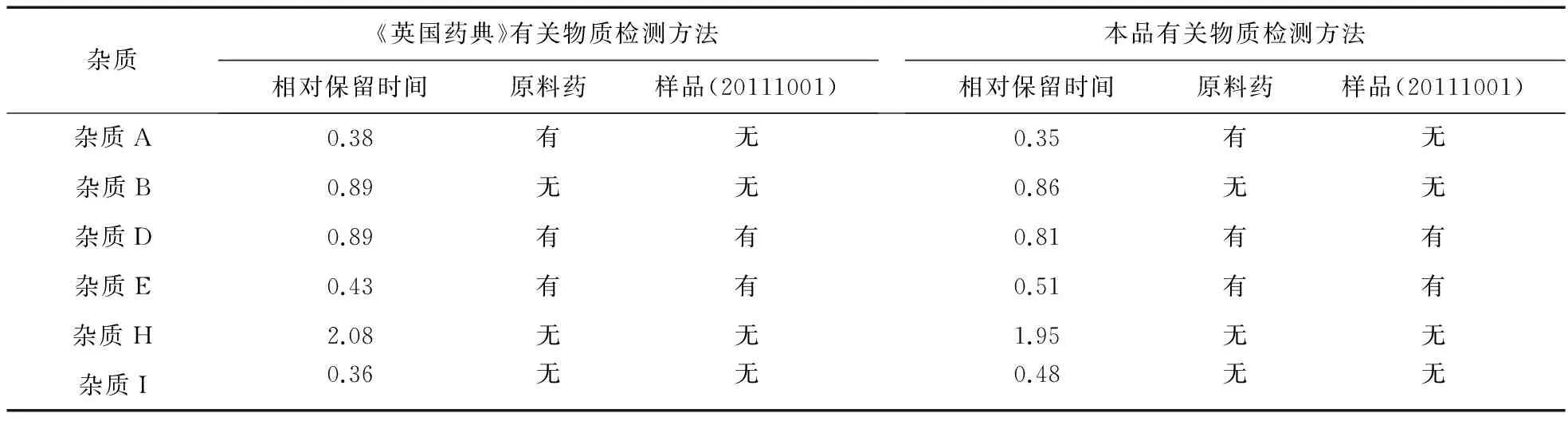
表4 杂质定性分析结果
[1] 李瑜元.质子泵抑制剂研发新进展[J].中国处方药,2006,(5):30-32.
[2] 国家药典委员会.中华人民共和国药典(二部)[S].北京:中国医药科技出版社,2010:1038-1040.
[3] 许国铭,方裕强,程能能,等.质子泵抑制剂(奥美拉唑)试验在胃食管反流病中的诊断价值[J].中华消化杂志,2002,22(1):7-10.
[4] 英国药典(一部)[S].奥美拉唑,2009:3
[5] 王旭,李美珍,卢欣,等.三种HPLC色谱条件下测定奥美拉唑含量方法的比较[J].天津医科大学学报,2012,18(4):510-513,517.
[6] 迟云峰,张明晶.复方奥美拉唑咀嚼片含量测定方法的研究[J].中国医药指南,2012,10(31):98-99.
[7] 孙蕊,刘世超.奥美拉唑镁肠溶片含量测定方法的改进[J].中国医药导报,2011,8(1):61-63.
[8] 王乐群,马长清.高效液相色谱法测定奥美拉唑肠溶胶囊的含量[J].中国药师,2008,11(6):656-657.
[9] 刘淑平,王东凯,高斐,等.RP-HPLC法测定注射用奥美拉唑钠中奥美拉唑及有关物质含量[J].沈阳药科大学学报,2006,23(6):369-372.
Detection of the content and related substances of compound omeprazole powder for oral suspension by RP-HPLC
TIAN Xiao-feng*,WANG Guo-jun
(Tieling Central Hospital,Tieling 112000,China)
Objective To establish an RP-HPLC method for the determination of omeprazole and its related substances in compound omeprazole powder for oral suspension.Methods A Phenomenex Luna C18column (250 mm×4.60 mm,5 μm) was used,the mobile phase consisted of methanol-water-triethylamine-phosphoric acid (V∶V∶V∶V=55∶45∶0.3∶0.12) at the rate of 1.0 mL/min,the column temperature was 30 ℃ and the injection volume was 20 μL;the detection wavelength was 302 nm and 280 nm for the determination of the content and related substances,respectively.Results Omeprazole was completely separated from its degradation products,the linear range was 0.133~30.0 mg/L(r=0.999 7),and the average recovery was 99.0%(RSD=0.2%,n=9).The contents of 3 batches of samples and reference drug were 97.5%,97.6%,97.5% and 92.3%,respectively.The maximum single impurity was 0.06%,0.06%,0.05% and 0.15%,respectively.The total impurities were 0.11%,0.11%,0.11% and 0.45%,respectively.Conclusion The method can be used for the determination of the content and related substances in compound omeprazole powder for oral suspension.
Omeprazole;HPLC;Content determination;Related substance
2016-08-26
铁岭市中心医院,辽宁 铁岭 112000
*通信作者
10.14053/j.cnki.ppcr.201704022
猜你喜欢
中国药学药品知识仓库(2022年13期)2022-07-03
人人健康(2021年14期)2021-08-06
艺术品鉴(2020年6期)2020-12-06
时代邮刊(2019年22期)2019-12-17
时代邮刊·下半月(2019年11期)2019-09-22
山东化工(2018年15期)2018-09-20
领导文萃(2017年6期)2017-03-24
中学生数理化·高一版(2016年7期)2016-12-07
首都食品与医药(2015年18期)2015-11-03
中学生数理化·中考版(2015年12期)2015-09-10
