
茶皂素的提取纯化及其单当归酰基茶皂苷元的制取
2017-04-25 02:05:55陈绪涛肖大瑾霍光华龙昊知刘景利江西农业大学生物资源保护和利用研究所生物科学与工程学院南昌330045
中国粮油学报 2017年3期
陈绪涛 肖大瑾 霍光华 龙昊知 刘景利(江西农业大学生物资源保护和利用研究所;生物科学与工程学院,南昌 330045)
茶皂素的提取纯化及其单当归酰基茶皂苷元的制取
陈绪涛 肖大瑾 霍光华 龙昊知 刘景利
(江西农业大学生物资源保护和利用研究所;生物科学与工程学院,南昌 330045)
榨取茶油后的茶籽饼粕含有丰富的茶皂素。为了精确定位茶皂素的生物活性及其构效关系,设计合成多样性药物分子,分离茶皂素单体和制取明确结构的苷元成为必要。本试验采用水浴提取,树脂吸附及沉析获得茶皂素;通过酸水解、溶剂萃取、柱层析和高效液相色谱等方法制备其茶皂苷元,色谱和波谱定性定量。结果显示,茶饼中茶皂素的水提取条件为:料液比1∶25(m/V)、80℃水浴,提取2 h,3次。纯化条件为:茶皂素水提取液过AB-8型树脂,依次用水、稀碱、水和90%乙醇洗脱,醇洗脱液冰浴沉析3 h,离心获得茶皂素,纯度达96%。然后将其进行盐酸水解,条件为:3 mol/L盐酸60%甲醇水溶液,料液比1∶15(m/V),85℃回流水解5 h;再进行乙酸乙酯萃取、硅胶柱层析,收集石油醚-乙酸乙酯洗脱液,液相制备,获得2种茶皂素苷元,波谱数据并与文献比对,确定其结构为:21-当归酰基-22-乙酰茶皂苷元Ⅰ和Ⅱ,两者仅有C4位甲基和醛基差别。
茶皂素 茶皂苷元 提取纯化
网络出版地址:http://www.cnki.net/kcms/detail/11.2864.TS.20170120.0853.002.html
茶(Camellia sp.)属山茶科(Theaceae)植物,其中油茶在我国种植面积现有5 000多万亩,并有逐年扩大的趋势,江西、湖南、广西等地为主产地[1]。油茶及其同属植物的种籽是榨取茶油的主要原料[2],相当于茶油三倍量的茶饼粕年产约40万t[3]。味苦有毒的茶饼粕,主要被用于肥料、燃料、清塘剂等[4],资源未能得到有效利用。茶饼粕含有10%左右[5]的混合茶皂素,可用于纺织业、养殖业、建材业、医药、日化、食品以及农业上[6],且提取茶皂素后的茶饼还可以作为有机肥料或者蛋白饲料等[7]。虽然已有许多学者开展过茶皂素的提取分离研究,也有市售茶皂素产品供应,但其中存在复杂的混合茶皂素和较多色素等杂质[8]。迄今人们已发现茶皂素种类至少有93种之多[9],混合茶皂素不利于活性精确定位和构效关系确认,也需要具有明确化学结构和足够产量的茶皂苷元来开展多样性药物分子设计和合成。
木荷叶皂苷具备强烈抗稻瘟病菌活性。彭玉萌等[10]制备出2种抗稻瘟病菌木荷皂苷,其EC50分别为5.91、5.20 mg/L,易磊等[11]研究发现木荷皂苷对稻瘟病菌细胞形态和生理生化指标具有较大的影响,但难以获得足够量的木荷皂苷苷元来实现其多样化的分子设计合成和构效关系研究。茶和木荷同属山茶科植物,均含丰富的当归酰基苷元的结构皂苷。此外,与木荷叶相比,茶饼含有干扰提取分离的色素较少等。因此,本试验研究茶饼粕茶皂素的提取纯化和其苷元的制备,以期为茶籽饼的开发利用提供参考。
1 材料与方法
1.1 材料
茶籽饼粕:南昌梅岭风景村后山榨油厂;齐墩果酸(白色粉末,≥95%):宝鸡市方晟生物开发有限公司;GF254层析板:青岛海洋化工厂分厂;硅胶(200~300目和300~400目):青岛海浪硅胶干燥剂有限公司;其他分析纯或色谱纯试剂:西陇化工。
1.2 仪器
EYELA旋转蒸发仪:上海爱朗仪器有限公司;JW-3022HR型高速冷冻离心机:安徽嘉文仪器装备有限公司;Sartorius电子自动分析天平:上海树信仪器仪表有限公司;KQ-C玻璃仪器气流烘干器:巩义市英峪予华仪器厂;KQ3200型超声清洗器:昆山市超声仪器有限公司;TU-1900型双光束紫外可见分光光度计:北京普析通用仪器有限责任公司;LC3000半制备型高效液相色谱仪:北京创新通恒科技有限公司;Waters 2695型高效液相色谱仪:美国Waters公司;Bruker400型核磁共振仪:瑞士Bruker公司。
1.3 方法
1.3.1 茶皂素的提取
称取60℃干制的茶饼粕粉,以料液比1∶10,80℃水浴,提取1 h,1次为基础,分别设定提取温度(40、50、60、70、80、90℃)、提取时间(15、30、60、120、180 min)、料液比(1∶5、1∶10、1∶15、1∶20、1∶25)、提取次数(1、2、3、4、5次)等单因素不同水平试验,每组试验重复3次,以茶皂素得率为指标。选取L27(313)正交设计表,进行四因素三水平正交试验并考察其交互作用,最后试验验证了最佳试验参数的提取效果。
1.3.2 茶皂素纯化
将茶皂素水提液直接过AB-8型大孔树脂[12],待饱和吸附后,依次用蒸馏水(洗至无色,后面同上)、0.1%NaOH、蒸馏水(洗至中性)冲洗,最后收集90%乙醇水洗脱液,冰浴3 h,离心获得纯化的茶皂素备用。
1.3.3 茶皂苷元制备[13]
以盐酸浓度为4 mol/L、水解温度为100℃、水解时间为6 h、料液比为1∶20(m/V)为基础,分别进行盐酸浓度(1、2、3、4、5 mol/L)、水解温度(60、70、80、90、100℃)、水解时间(3、4、5、6、7 h)和料液比(1∶5、1∶10、1∶15、1∶20、1∶25)等单因素试验后,选用L9(34)正交设计进行正交试验获得优化的茶皂苷元制备参数并验证最佳参数组合,再用石油醚-乙酸乙酯(3 ∶1、2∶1、1∶1)为洗脱液,分别以200~300目和300~400目为分离材料,过硅胶柱,半制备高相液相色谱制得茶皂苷元单体[色谱条件:BDS HYPERSIL C18色谱柱(250 mm×10 mm,5 μm),流动相A为色谱纯乙腈,B为二次蒸馏水,检测波长为280 nm,柱温30℃,流速2 mL/min,进样量100 μL,记录时间30 min,洗脱程序为均相洗脱即A相72%、B相为28%]。
1.3.4 茶皂素及其皂苷元的定性定量分析
1.3.4.1 薄层(TLC)定性检测
采用二元展开剂展开,茶皂素展开剂为氯仿∶甲醇=7∶10;茶皂苷元展开剂为石油醚∶乙酸乙酯= 1∶1。显色剂均为10%硫酸乙醇溶液[14]。
1.3.4.2 高效液相色谱(HPLC)分析茶皂素及其苷元
准确称取水提粗茶皂素、纯化茶皂素及其茶皂苷元各2 mg,溶于1 mL色谱纯甲醇超声溶解,0.45 μm微孔滤膜过滤。利用Waters分析型高效液相进行分析。色谱条件:BDS HYPERSIL C18色谱柱(250 mm×4.6 mm,5 μm);流动相A为色谱纯乙腈,B为二次蒸馏水,检测波长为280 nm,柱温30℃,流速0.5 mL/min,进样量5 μL,记录时间1 h,茶皂素洗脱程序为0~25~35~40~55~60 min,10% ~60% ~62%~78%~80%~100%(A相),混合茶皂苷元洗脱程序为0~3~10~25~30 min,40% ~60%~90% ~100%(A相),单体茶皂苷元洗脱程序为均相洗脱即A 相72%、B相为28%。
1.3.4.3 比色法测定茶皂素含量
分别称取提取的粗茶皂素、纯化茶皂素和齐墩果酸各0.015 0 g,溶于甲醇并定容至50 mL,准确称量香草醛0.506 1 g,溶于冰醋酸并定容至10 mL,备用。分别取已配好的齐墩果酸母液0、0.2、0.4、0.6、0.8、1.0 mL于6支干净的试管中,水浴蒸干,再向每支试管加0.8 mL高氯酸、0.2 mL已配好的香草醛,70℃水浴30 min,然后冰浴5 min后向各试管加入5 mL冰醋酸,550 nm处测定吸光值[15]。以吸光度值对齐墩果酸的质量浓度绘制标准曲线,得到:Y= 5.750 1X-0.006 9(R2=0.997 5),说明齐墩果酸在10~50 μg/mL与吸光度呈现很好的线性关系。同样测定样品的吸光度值,根据此曲线计算出相应浓度。
1.3.4.4 显色法[14]和核磁(NMR)波谱法判别茶皂苷元的结构
Molish反应:取2 mg茶皂苷元粉末于试管中,加3 mL甲醇溶解后,加入2滴10% α-萘酚乙醇溶液,摇匀,沿试管壁加入1 mL浓硫酸,观察界面处颜色变化;Libermann Burchard反应:取2 mg茶皂苷元粉末于试管中,加2 mL醋酸酐溶解后,加2滴浓硫酸摇匀,观察混合液颜色变化。
取10mg纯化的茶皂苷元用CD3OD溶解,转入NORELL-ST500核磁管中,上 NMR波谱仪测定。将其碳、氢谱化学位移和偶合常数等与文献比对和分析,确定了茶皂苷元的结构。
2 结果与分析
2.1 茶皂素提取参数优化
2.1.1 单因素对茶皂素提取率的影响
由图1可知,随着提取温度的提高茶皂素提取率也在升高,但是在80℃后有所下降;提取3次之后,茶皂素提取率没有很大变化;提取时间在2 h前,茶皂素提取率随时间而增加,2 h后提取率趋于平缓;料液比亦有类似规律,即当料液比在1∶15前,茶皂素提取率随料液比增加而快速提高,在其之后,随着料液比增加而提取率增加趋于缓慢变化。
2.1.2 多因素及其交互作用对茶皂素提取率的影响
表1中,茶皂素提取率的极差分析结果显示,4个因素对茶皂素提取率的影响能力大小为:提取次数(B)>料液比(D)>提取温度(A)>提取时间(C)。优化的茶皂素提取条件为:A3B3C1D3,即提取温度为90℃,提取次数为4次,提取时间为1 h,料液比为1∶25。
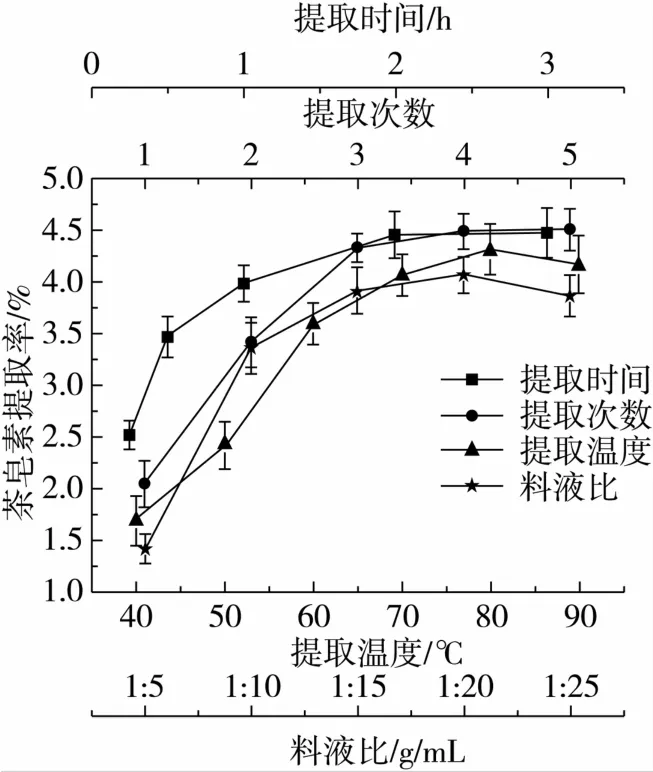
图1 不同因素对茶皂素提取率的影响

表1 茶皂素提取正交试验结果
方差分析见表2。由F分布临界值表查F0.01(2,6)=10.92,F0.05(2,6) =5.14,F0.01(4,6)=9.15,F0.05(4,6) =4.53,取显著性水平为0.05。因素A、B、C、D的F值和交互作用(A×B)、(C×D)、(A× D)、(B×D)的F值分别与临界值F0.05(2,6)和F0.05(4,6)对比。因素B、D的F值远远大于5.14,因素(A×B)、(A×D)、(B×D)的F值远远大于4.53,对试验结果有显著的影响。因素A、C、(C×D)、(A× C)对试验结果具有一定的影响。各试验因素对茶皂素提取率的影响程度大小依次为:提取次数(B)>料液比(D)>(B×D)>(A×B)>(A×D)>(C×D)>提取温度(A)>提取时间(C)>(A×C)。由方差可以看出,应优先确定提取次数(B)和料液比(D),而提取温度(A)根据(A×B)交互作用来确定,提取时间(C)则根据(C×D)交互作用来确定,(A×B)和(C×D)交互搭配由表3,表4可得,提取温度为A2即80℃时茶皂素提取率最高,提取时间为C3即3 h时茶皂素提取率最高,则最优搭配方案为A2B3C3D3。
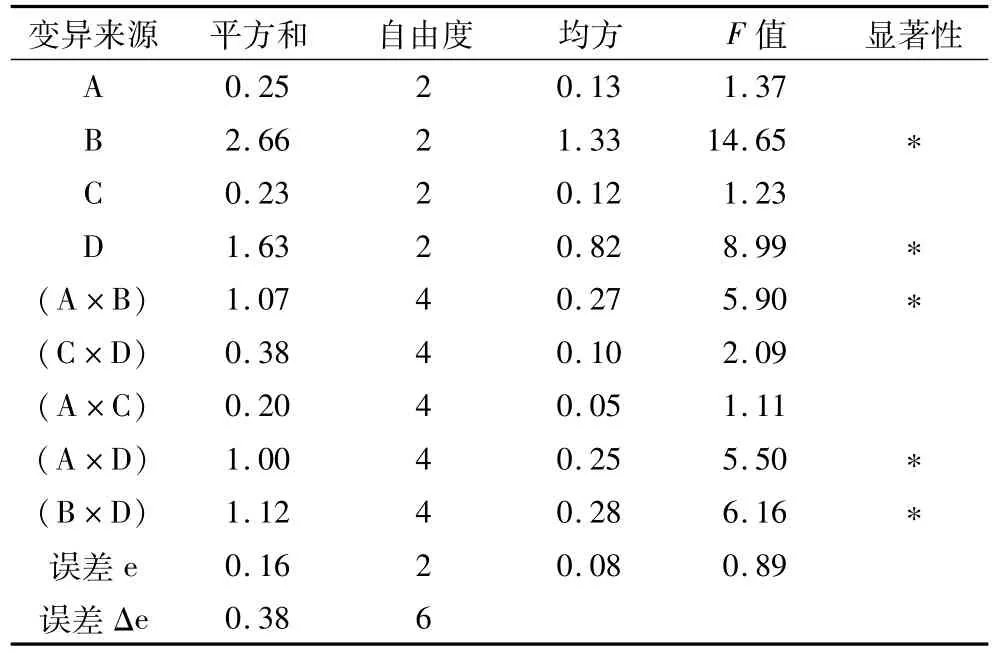
表2 茶皂素提取率方差分析
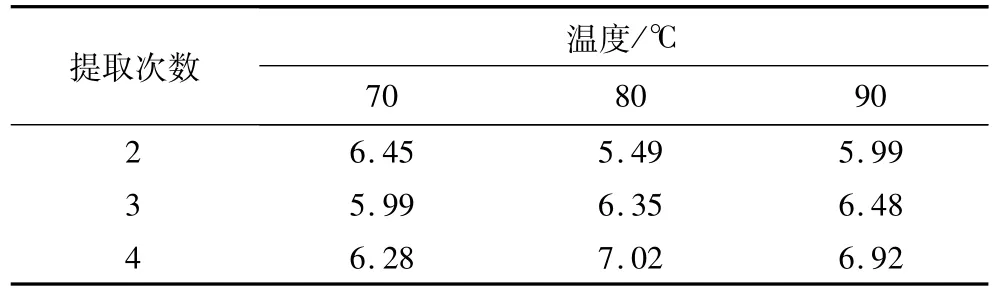
表3 不同提取次数不同提取温度茶皂素的提取率/%

表4 不同料液比不同提取时间的茶皂素提取率/%
综上所述,提取次数和料液比存在最优值B3、D3,而提取温度与提取时间在茶皂素提取率分析中都归于误差,其最优水平由交互作用确定。按照该两组最优条件即A3B3C1D3和A2B3C3D3进一步考察茶皂素提取率,其结果分别为(7.83±0.13)%和(8.12±0.17)%。所以水提茶籽饼粕中茶皂素的理论最优条件取:A2B3C3D3,即 80℃水浴,料液比1∶25,每次3 h,提取4次。考虑到实际耗能等经济问题,并且在单因素试验中可以看到提取次数B为3次和4次时对结果影响不大,提取时间C为2 h和3 h时也对结果没有显著影响,结合理论最优条件,将因素 B2、C2进行试验考察,即组合 A2B2C3D3、A2B3C2D3、A2B2C2D3,经过试验验证,A2B2C3D3、A2B3C2D3、A2B2C2D33种组合茶皂素的提取率为(8.05±0.21)%、(8.07±0.16)%、(8.01±0.14)%。因此,最少耗能的组合A2B2C2D3的茶皂素提取率与理论最优组合的提取率相差不大,故选取A2B2C2D3,即80℃水浴,料液比1∶25,每次2 h,提取3次。
2.2 茶皂苷元的制备
2.2.1 单因素对茶皂素水解的影响
单因素试验结果显示在盐酸浓度为3 mol/L、水解温度为80℃、水解时间为5 h、料液比为1∶20 (m/V),各自茶皂苷元得率最高。

图2 不同因素对茶皂苷元得率的影响
2.2.2 多因素对茶皂素水解的影响
表5极差分析结果可以看出,各因素对茶皂素水解影响的主次顺序为A(盐酸浓度)>B(水解温度)>C(水解时间)>D(料液比),故茶皂素盐酸水解的最佳工艺为A2B3C2D1,即盐酸浓度为3 mol/L、水解温度为85℃、水解时间为5 h、料液比为1∶15 (m/V)。依此参数组合验证茶皂苷元平均得率为(7.89±0.22)%。

表5 茶皂素水解正交试验[L9(3)4]结果
2.3 提取纯化的茶皂素和水解纯化后的茶皂苷元
2.3.1 茶皂素及其苷元的薄层色谱(TLC)
如图3所示:茶皂素因极性较大,在极性较弱展层剂即石油醚∶乙酸乙酯体系展开时,几乎无上移(a,b),而在极性较强展开剂即氯仿∶甲醇体系展开时,粗品和纯化后的茶皂素比移值分别为 Rf= 0.35,0.68(d)和Rf=0.35(e);茶皂苷元因极性较其茶皂素减弱,在极性弱和极性强展层剂中比移值分别为Rf=0.43(c)和Rf=0.88(f);不同纯化程度的茶皂苷元在极性弱的展层剂中显示不同的斑点,即粗茶皂苷元(g)显一条长带,过一次柱后(i)是椭圆形带,主点Rf=0.43,而过二次柱纯化的仅有1个点即Rf=0.43的小椭圆点(h)。

图3 茶皂素水解前后薄层图
2.3.2 茶皂素及其苷元高效液相色谱(HPLC)
比色法测定出纯化获得的茶皂素纯度为96%,粗品茶皂素、纯化茶皂素和茶皂苷元HPLC色谱图如图4。粗品茶皂素出峰保留时间大致有3个时间段,即1~4 min,7~9 min,12~17 min;纯化茶皂素出峰保留时间主要在12~17 min,在其余少数时间有少量小峰出现。由此,茶皂素混合皂苷在该色谱程序下出峰保留时间为12~16 min,不难看出杂质色素等出峰保留时间在2~4 min和7~9 min,其中保留时间在7~9 min的为大部分杂质类物质;纯化后茶皂苷元在梯度洗脱中,保留时间分别为15.8 min和19.2 min,单体茶皂苷元Ⅰ和Ⅱ在等度洗脱中,保留时间分别为5.1 min和6.2 min。

图4 茶皂素及其茶皂苷元的高效液相色谱图
2.3.3 制备的茶皂苷元的结构
茶皂素酸解产物Molish反应呈现阴性,即与浓硫酸接触面处无紫色环出现,说明无糖基的存在。LibermannBurchard反应呈现阳性,即出现颜色有红紫蓝绿的变化,说明该物质含有皂苷元骨架。
茶皂苷元Ⅰ碳、氢核磁共振谱化学位移:1HNMR(400MHz,CD3OD)δ 0.78(3H,s,25-H3),0.90(3H,s,26-H3),0.92(3H,s,29-H3),0.95(3H,s,30-H3),0.99(3H,s,24-H3),1.83 (3H,d,27-H3),1.69(3H,d,J=7.4Hz,21-OAng-4-H3),1.87(3H,s,22-O-Ac),1.99 (3H,s,21-O-Ang-5-H3),3.23(1H,dd,18 -H),3.50,3.60(2H,d,J=11.5Hz,28-H2),4.08(1H,m,3-H),4.34(1H,brs,16H),5.37 (1H,brs,12-H),5.84(1H,d,J=11.2Hz,22-H),6.87(1H,d,J=11.2Hz,21-H),7.4(1H,s,21-O-Ang-3-H),9.29(1H,s,23-H).13CNMR(101 MHz,CD3OD)δ 9.6(C24),12.8(21-O -Ang,C5′),14.6(21-O-Ang,C4'),15.4 (C25),17.4(C26),21.0(C30),21.8(C6),24.7 (22-O-Ac,C2″),25.6(C11),27.1(C2),27.8 (C27),28.9(C29),32.2(C7),33.7(C15),37.0 (C10),38.2(C20),39.7(C1),41.6(C18),42.6 (C8),44.7(C14),44.9(C9),45.4(C19),48.1 (C17),48.3(C5),56.9(C4),61.7(C28),70.19 (C16),72.9(C22),73.8(C21),79.8(C3),125.1 (C12),130.9(21-O-Ang,C2′),139.6(21-OAng,C3′),143.0(C13),168.8(21-O-Ang,C1′),173.1(22-O-Ac,C1″),208.6(C23)。
茶皂苷元Ⅱ碳、氢核磁共振谱化学位移:1HNMR(400MHz,CD3OD)δ 0.71(3H,s,25-H3),0.82(3H,s,26-H3),0.90(3H,s,29-H3),0.93(3H,s,30-H3),1.00(3H,s,24-H3),1.82 (3H,d,27-H3),1.91(3H,d,J=7.6Hz,21-O -Ang-4-H3),2.18(3H,s,22-O-Ac),2.40 (3H,s,21-O-Ang-5-H3),3.14(1H,dd,18 -H),3.87,3.92(2H,d,J=11.5Hz,28-H2),4.06(1H,m,3-H),4.91(1H,brs,16H),5.24 (1H,brs,12-H),5.77(1H,d,J=11.1Hz,22-H),6.86(1H,d,J=11.2Hz,21-H),7.20(1H,s,21-O-Ang-3-H).13C-NMR(101 MHz,CD3OD)δ 13.9(C24),15.3(21-O-Ang,C5'),16.8(21-O-Ang,C4'),18.1(C25),20.8 (C26),23.2(C30),25.6(C6),26.9(22-OAc,C2"),27.4(C11),27.8(C2),29.4(C27),31.6(C29),32.5(C7),33.1(C15),34.8 (C10),36.7(C20),38.3(C1),38.5(C18),39.7(C8),40.2(C14),41.7(C9),42.9(C19),45.8(C17),46.4(C5),54.9(C4),69.9(C28),71.2(C16),73.1(C22),74.6(C21),77.9 (C3),123.1(C12),129.7(21-O-Ang,C2'),138.0(21-O-Ang,C3'),141.4(C13),169.2(21 -O-Ang,C1'),177.9(22-O-Ac,C1")。
Ⅰ其中δC168.8、δH6.87(1H,d)和δC173.1、δH5.84(1H,d)分别为C21位的当归酰基和C22位的乙酰基,δC208.6和 δH9.29为 C23位的醛基。δC125.1和 δC143.0则为 C12、C13位的双键。δc 61.7、δC70.1、δC79.8分别反映出C28位伯羟基、C16位和 C3位仲羟基,最后 δc 9.6~δC 56.9和δH0.78(3H,s)~δH5.37(1H,brs)则体现出齐墩果烷型五环骨架。Ⅱ其中δC169.2、δH6.86(1H,d)和δC177.9、δH5.77(1H,d)分别为C21位的当归酰基和C22位的乙酰基。δC123.1和δC141.4则为C12、C13位的双键。δc 69.9、δC71.2、δC77.9分别反映出C28位伯羟基、C16位和C3位仲羟基,最后δc13.9~δC 54.9和 δH0.71(3H,s)~δH5.24 (1H,brs)则体现出齐墩果烷型五环骨架,上述数据均与文献[16-18]一致,故确定所制备的茶皂苷元的结构为:21-当归酰基-22-乙酰茶皂苷元Ⅰ和Ⅱ,两者仅有C4位甲基和醛基差别。结构式如图5。
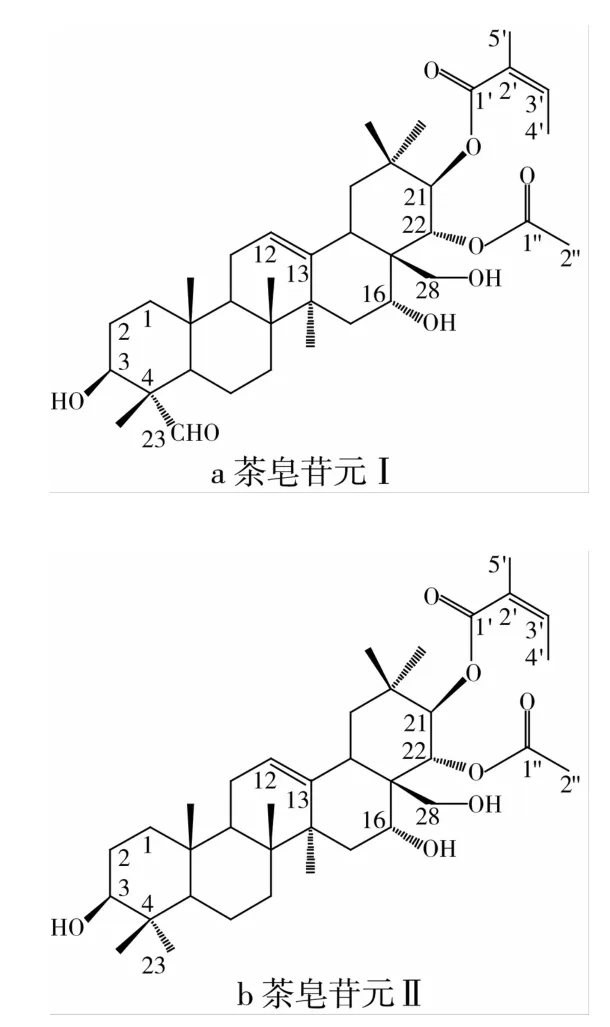
图5 21-当归酰基-22-乙酰茶皂苷元结构式
3 讨论与结论
水提取茶饼粕中的茶皂素,最佳提取参数:料液比1∶25(m/V),80℃水浴提取2 h,重复3次,总茶皂素得率为8.01%。经AB-8大孔树脂和冰浴沉析纯化获96%的混合茶皂素。茶皂苷元制备的水解条件为:3 mol/L盐酸,料液比为1∶15(m/V)的60%甲醇水溶液85℃加热回流5 h,分离制备获得21-当归酰基-22-乙酰茶皂苷元Ⅰ和Ⅱ,两者仅有C4位甲基和醛基差别。
用水提取茶皂素,相对于醇提取茶皂素成本低,且有8.01%得率,与谢多等[19]乙醇提取茶皂素得率8.34%、彭游等[20]微波提取茶皂素9%相差不大。马力等[21]在料液比1∶11、pH=9、80℃水浴6 h可萃取出茶饼中95.5%的茶皂素,但是在高温碱性条件下可能会破坏茶皂素部分结构,特别是其结构中的酯键[22]。
AB-8大孔树脂和冰浴沉析纯化能有效的去除混合茶皂素中的色素杂质,茶皂素所获纯度96%比H2O2高温碱性条件脱色87.4%[23]更高。
茶皂素苷元种类至少有51种以上[5],主要结构是齐墩果烷骨架,区别是当归酰基与惕各酰基在C21 和C22变换,羟基(包括伯羟基)和乙酰基在C15、C16、C28位变换,醛基、甲基和伯羟基在 C23位变换。本试验分离制备出其中2种,C21位当归酰基和C22位乙酰基的茶皂苷元,与前期鉴定的木荷玉蕊醇苷元[24]极其相似,较方便的提供了含一个当归酰基苷元结构,满足了多样化含当归酰基齐墩果烷型苷元糖基化合成的需要,奠定了其构效关系研究的基础和合成多样化皂苷分子的需要。
[1]张超英.谈我国油茶产业的发展[J].国家林业局管理干部学院学报,2013,12(1):21-24 Zhang C Y.Discussion on the development of Chinese oil camellia industry[J].State Academy of Forestry Administration Journal,2013,12(1):21-24
[2]Chen Y F,Yang C H,Chang M S,et al.Foam properties and detergent abilities of the saponins from camellia oleifera [J].Molecules,2010,11(11):4417-25
[3]杨强,胡海波,张石蕊.茶粕饲料资源开发及利用技术研究进展[J].饲料工业,2006,27(19):53-55 Yang Q,Hu H B,Zhang S R.Advances on the study of tea meal feed resources development and utilization technology [J].Feed Industry,2006,27(19):53-55
[4]王建枫,程晓建.油茶籽粕饲料资源的开发与利用[J].饲料研究,2015(10):65-68 Wang J F,Cheng X J.Tea seed meal feed resources development and utilization[J].Feed Research,2015(10):65-68
[5]Zhao P,Gao D F,Xu M,et al.Triterpenoid Saponins from the Genus Camellia[J].Chemistry and Biodiversity,2011,8(11):2881-5
[6]刘晓仙,何杨林,刘芳珍.茶皂素的应用综述[J].蚕桑茶叶通讯,2007(2):24-25 Liu X X,He Y L,Liu F Z.Review about the application of tea saponin[J].Newsletter of Sericulture and Tea,2007(2):24-25
[7]刘玲玲,李庆松,曾亮,等.茶籽饼中茶皂素提取工艺研究[J].粮食与食品工业,2010,17(4):19-22 Liu L L,Li Q S,Zeng L,et al.Research on extracting technology of tea saponin from tea-seed cakes[J].Cereal and Food Industry,2010,17(4):19-22
[8]袁华,刘瑞华,张能敏.一种提纯粗茶皂素的简易方法[J].生物加工过程,2008,6(5):18-20 Yuan H,Liu R H,Zhang N M.A simple method for purification of crude tea saponin[J].Chinese Journal of Bioprocess Engineering,2008,6(5):18-20
[9]陈瑶,杨文,周玉锋.茶皂素对植物病原菌活性研究进展[J].贵州茶叶,2014(1):1-5 Chen Y,Yang W,Zhou Y F.Research progresses on antiphytopathogen activity of tea saponin[J].Journal of GuiZhou Tea,2014(1):1-5
[10]彭玉萌,霍光华,韩启灿,等.抗稻瘟病菌活性木荷皂甙类似物的分离条件及其分离[J].分析化学,2014,42 (1):59-64 Peng Y M,Huo G H,Han Q C,et al.Separation of saponin analogue from Schima Superba with activity resisting Magnaporthe Oryzae[J].Chinese Journal of Analytical Chemistry,2014,42(1):59-64
[11]易磊,霍光华,韩启灿,等.木荷皂甙对稻瘟病菌细胞形态及生理生化指标的影响[J].植物保护学报,2013,40(5):450-456 Yi L,Huo G H,Han Q C,et al.Effects of saponins from Schima superba on cell morphology,physiological and biochemical indices of Magnaporthe oryzae[J].Journal of Plant Protection,2013,40(5):450-456
[12]赵娟,黄健花,王兴国.树脂法纯化油茶皂素的工艺研究[J].农业机械,2011(8):143-147 Zhao J,Huang J H,Wang X G.Purification of sasanqua saponin by resin process research[J].Farm Machinery,2011(8):143-147
[13]王延芳.油茶皂素及其水解产物的分离及降血脂抗氧化活性研究[D].广州:华南理工大学,2012 Wang T F.Studies on separation of sasanquasaponin and its hydrolyzed products and their hypolipidemic and antioxidant activities[D].Guangzhou:South China University of Technology,2012
[14]李静,李燕,党培育.茶皂素的提取及纯化研究[J].食品科学,2008,29(11):154-156 Li J,Li Y,Dang P Y.Study on extraction and purification of tea saponin[J].Food Science,2008,29(11):154-156
[15]干丽,何桂霞,李嘉滢,等.茶枯饼中茶皂素苷元提取工艺的研究[J].中南药学,2014(12):1199-1201 Gan L,He G X,Li J Y,et al.Extraction technology of tea saponin aglycone from tea oil cakes[J].Central South Pharmacy,2014(12):1199-1201
[16]Masayuki Y,Toshio M,Ning L,et al.Bioactive Saponins and Glycosides.ⅩⅩⅢ.Triterpene Saponins with Gastroprotective Effect from the Seeds of Camellia sinensis-Theasaponins E3,E4,Es,E6,and E7[J].Chemical& Pharmaceutical Bulletin,2005,53(12):1559-1564
[17]Masayuki Y,Toshio M,Seikou N,et al.Bioactive Saponins and Glycosides.ⅩⅩⅤ.Acylatedoleanane-TypeTriterpeneSaponins from the Seeds of Tea Plant(Camellia sinensis)[J].Chemical and Pharmaceutical Bulletin,2007,55(1):57-63
[18]Chen J H,Wu H Y,Liau B C,et al.Identification and evaluation of antioxidants defatted Camellia oleiferaseedsby isopropanol salting-out pretreatment[J].Food Chemistry,2010,121(4):1246-1254
[19]谢多,赵俭,许小彤,等.油茶饼粕中茶皂素醇提工艺条件优化[J].食品与机械,2013(3):129-133 Xie D,Zhao J,Xu X T.et al.Optimization of alcohol extraction for tea saponin from oil-tea cake by response surface methodology[J].Food and Machinery,2013(3):129-133
[20]彭游,朗少杰,邓泽元,等.茶皂素光波干法提取与产品开发[J].天然产物研究与开发,2009,21(6):1023-1027 Peng Y,Lang S J,Deng Z Y,et al.Tea saponin extraction by light wave without solvent and its application[J].Natural Product Research and Development,2009,21(6):1023-1027
[21]马力,陈永忠,彭邵锋,等.利用水作溶剂提取油茶粕中茶皂素的工艺研究[J].农业科学与技术:英文版,2015 (5):1078-1080 Ma L,Chen Y Z,Peng S F,et al.Study on Extraction of Tea Saponin from Camellia oleifera Cake Using Water as Extraction Solvent[J].Agricultural Science and Technology,2015(5):1078-1080
[22]胡健华,陈新新.油茶皂素、油茶多糖及糖萜素的化学结构、理化性质综述[J].武汉工业学院学报,2012(2):20-23 Hu J H,Chen X X.The review of the chemical structure,physical and chemical properties on the tea saponin,tea polysaccharide and saccharicterpenin[J].Journal of Wuhan Polytechnic University,2012(2):20-23
[23]王超,仲山民,郑旭卫.油茶饼粕中茶皂素的提取及H2O2法脱色条件研究[J].粮油加工,2009(6):71-73 Wang C,Zhong S M,Zhen X W.Study on the extraction of tea saponin and H2O2decolorization conditions of oil-tea cake[J].Cereals and Oils Processing,2009(6):71-73
[24]Huo G H,Zhang C L,Zhang Y J.Structure Elucidation of TwoTriterpenoidSaponins from Leaves of SchimasuperbaGardn.et Champ[J].Lecture Notes in Electrical Engineering,Springer,2014,250(2):915-922.
Extraction and Purification of Tea Saponin and Preparation of Single Angeloyl theasapogenol
Chen Xutao Xiao Dajin Huo Guanghua Long Haozhi Liu Jingli
(Institute of Bioresource Conservation and UtilizationCollege of Bioscience and Engineering,Jiangxi Agricultural University,Nanchang 330045)
Tea seed cake is the by-product of tea seed oil extracted and rich in tea saponin.In order to locate accurately its relationship between biological activity and structure-activity,design and synthesis of multifarious molecular drug,separation of tea saponin monomer and preparation of theasapogenol with explicit structure become necessary.In this paper,tea saponins were obtained using water bath extraction,resin adsorption and precipitation.The theasapogenols were prepared using acid hydrolysis,solvent extraction,column chromatography and HPLC.Qualitative and quantitative analysis of theasapogenol was performed by chromatography and spectrum.The results showed that the conditions for the water extraction of tea saponins were:ratio of material to liquid:1∶25(m/V),under waterbath at 80℃ for 2 h,triplicate.Purification conditions were as follows:Tea saponins were purified by AB-8 resin with elution H2O,0.1%NaOH,H2O and 90%ethanol successively,then precipitated 3 h,centrifuged and obtained with purity 96%.Two theasapogenols were prepared by the process of acid hydrolysis in 3 mol/L 60%HCl-Methanol aqueous solution with the ratio of material to liquid 1∶15(m/V),reflux at 85℃ for 5 h;extract with ethyl acetate,elution through silica gel column with petroleum ether-ethyl acetate,and semi-preparative HPLC.The theasapogenols were identified as 21-O-angeloyl-22-O-acetyl theasapogenolⅠandⅡaccording to their spectrum data and compared with literatures.TheasapogenolⅠdiffers from theasapogenolⅡonly at the group C-4 position,4-aldehyde or 4-methyl.
tea saponin,theasapogenol,extraction and purification
TS229
A
1003-0174(2017)03-0088-09
时间:2017-01-20 08:53:13
国家自然科学基金(21266010),江西省研究生创新专项资金(YC2014-S196),江西省自然科学基金(20132BAB204028)
2015-12-31
陈绪涛,男,1990年出生,硕士,微生物学
霍光华,男,1963年出生,博士,教授,天然药用成分鉴定、制备与产品开发
猜你喜欢
农药科学与管理(2019年10期)2019-04-20 07:13:14
天然产物研究与开发(2018年1期)2018-02-02 07:21:24
中国洗涤用品工业(2017年2期)2017-04-16 05:07:45
新农业(2016年13期)2016-08-16 12:12:42
当代化工研究(2016年2期)2016-03-20 16:21:23
中国洗涤用品工业(2016年2期)2016-02-28 19:03:17
中国粮油学报(2016年1期)2016-02-06 02:17:04
中国粮油学报(2016年1期)2016-02-06 02:16:54
应用化工(2014年5期)2014-08-08 13:10:58
食品科学(2013年17期)2013-03-11 18:27:13
