
前列腺癌差异表达基因的筛选及相互作用的生物信息学分析
2017-04-14 02:11夏前林单孟林朱延军郑江花
中国癌症杂志 2017年3期
夏前林,单孟林,丁 滔,朱延军,侯 君,郑江花,5
1.复旦大学附属公共卫生临床中心医学检验科,上海 201508;
2.上海交通大学附属第六人民医院南院泌尿外科,上海 201499;
3.复旦大学附属中山医院泌尿外科,上海 200032;
4.复旦大学附属中山医院病理科,上海 200032;
5.复旦大学附属公共卫生临床中心感染控制科,上海 201508
前列腺癌差异表达基因的筛选及相互作用的生物信息学分析
夏前林1,单孟林1,丁 滔2,朱延军3,侯 君4,郑江花1,5
1.复旦大学附属公共卫生临床中心医学检验科,上海 201508;
2.上海交通大学附属第六人民医院南院泌尿外科,上海 201499;
3.复旦大学附属中山医院泌尿外科,上海 200032;
4.复旦大学附属中山医院病理科,上海 200032;
5.复旦大学附属公共卫生临床中心感染控制科,上海 201508
背景与目的:基因芯片技术是利用杂交测序方法,可大规模高通量地检测不同组织、细胞中的基因表达水平的技术。该研究采用基因芯片技术筛选前列腺癌及前列腺炎症穿刺组织中差异表达的基因并对其进行生物学信息分析。方法:采用美国Affymetrix Human U133 plus 2.0基因表达谱芯片,按一步法分别抽提恶性程度较高的前列腺癌及前列腺炎症组织的总RNA,并分离纯化这两种组织的mRNA。经逆转录合成掺入生物素标记的cDNA合成探针,与芯片杂交和严格洗片后,用荧光扫描仪扫描芯片荧光信号图像。用生物信息学方法分析前列腺癌及前列腺炎症组织中差异表达基因。结果:按照表达差异倍数大于等于2,P<0.05的筛选条件,筛选出差异基因1 819条,其中上调表达基因1 025条和下调表达基因794条。采用GO富集分析发现这些差异基因主要涉及细胞周期、分子代谢等分子功能以及生物学过程;KEGG信号通路分析发现,这些差异基因主要涉及嘌呤核苷酸代谢等代谢通路。STING在线工具分析对差异基因编码的蛋白质间的相互作用进行分析,结果发现这些基因编码的蛋白间的相互作用主要集中在TPX2、ANLN、NUSAP1、MELK、DLGAP5、KIF11、TOP2A及RRM2等20个关键节点基因。最后对重要节点基因进行文献挖掘,发现CEP55和ANLN基因可能与前列腺癌的发生和转移有关。结论:在前列腺癌的发生、发展中,促使细胞进入增殖周期的相关基因的活化、代谢相关酶类基因活性的异常、细胞黏附功能相关基因的抑制以及细胞移行相关基因的激活等因素发挥了重要的作用。系统的分析这些基因发现,CEP55和ANLN基因与前列腺癌的发生和预后关系密切,为下一步研究实验提供有价值的线索。进一步研究相关差异基因将有望发现新的早期诊断指标,为建立个体化治疗方案和预后评估系统提供帮助。
前列腺癌;基因芯片;差异基因;生物信息学
前列腺癌在许多欧美国家是男性最常见的恶性肿瘤之一,其死亡率仅次于肺癌,居男性肿瘤的第二位[1]。虽然我国前列腺癌的发病率较低,但随着我国人口老龄化、饮食结构调整及诊断技术的提高,该发病率逐年增高,跃居男性泌尿生殖系统恶性肿瘤的第3位。基因芯片技术是将大量的靶基因全长或片段有序而高密度地固定排列在玻片、硅片或尼龙膜固相支持物上的一项技术,具有高通量、多参数、平行化等优点,在功能基因组学研究中正发挥越来越重要的作用。自问世以来基因芯片技术已广泛用于人类多种肿瘤基因表达谱等方面的研究,为肿瘤相关基因的研究提供了一种全新的方法。目前,利用基因芯片进行前列腺癌的研究主要集中在三个方面,包括通过分析前列腺癌以及正常前列腺组织进行前列腺癌发生、发展相关的基因研究[2],通过分析原发性前列腺癌组织和转移性前列腺癌组织进行前列腺癌侵袭和转移相关的基因研究[3]以及通过分析治疗后复发和未复发的前列腺癌患者组织标本进行前列腺癌预后和复发倾向预测的研究[4]。虽然,利用基因芯片筛选前列腺癌差异基因的报道较多,但对前列腺癌与前列腺炎症组织差异表达基因分析的报道甚少。因此,有必要对相关的差异表达基因进行筛选分析。本实验应用基因芯片技术检测了恶性程度较高的前列腺癌和前列腺炎症穿刺组织中基因表达的不同,初步筛选出相关的基因,为进一步深入研究其在前列腺癌的发生、发展中的作用以及找到更客观的临床诊断指标提供新的线索。
1 材料和方法
1.1 材料
1.1.1 基因芯片
所用基因芯片为美国Affymetrix公司的U133 plus 2.0基因表达谱芯片,由上海伯豪生物技术有限公司提供。该芯片包含了人类基因组47 000个转录本,涵盖了38 500个明确的人类基因。其芯片数据库来源于GenBank、dbEST和RefSeq。序列簇信息由UniGene(2003年1月25日)数据库建立,并经由华盛顿大学、加州大学圣克鲁斯分校和美国国立生物技术信息中心(National Center for Biotechnology Information,NCBI)的公共数据库共同整合而成。
1.1.2 前列腺癌组织标本
按照我国医学伦理委员会制度与操作规程进行临床标本的资料和收集。4例前列腺癌组织及4例前列腺炎症组织穿刺标本来自于上海市复旦大学附属中山医院泌尿外科,均按照2014年《中国前列腺癌诊断治疗指南》经组织病理学检查确诊。患者年龄57~73岁,平均65岁。将标本收集后迅速置于-80 ℃冰箱冷冻保存待检。
1.2 方法
1.2.1 前列腺组织RNA的提取和纯化
前列腺炎症组织和前列腺癌组织总RNA的提取采用TRIzol一步法抽提,用超微量分光光度计NanoDrop ND-2000在260 nm和280 nm处分别测定吸光度值(D),抽提所得总RNA经Agilent Bioanalyzer 2100(购自美国Agilent科技有限公司)电泳质检合格后使用RNeasy迷你试剂盒(型号:74004,购自德国QIAGEN公司)和RNase-Free DNase Set(型号:79254,购自德国QIAGEN公司)纯化总RNA。
1.2.2 样品RNA的放大和标记
实验样品RNA采用Affymetrix表达谱芯片配套试剂盒GeneChip 3’ IVT PLUS反转试剂盒(型号:902416,购自美国Affymetrix公司)对样品总RNA中的mRNA进行放大、标记和纯化,获得带有生物素标记的cRNA。
1.2.3 芯片杂交
按照Affymetrix表达谱芯片配套提供的杂交标准流程和配套试剂盒GeneChip® 杂交清洗染色试剂盒(型号:900720,购自美国Affymetrix公司)说明书进行操作。在滚动杂交炉中在45 ℃的条件下滚动杂交16 h,杂交完成后把杂交好的芯片进行洗脱、染色。
1.2.4 芯片扫描和检测分析
芯片杂交结果采用GeneChip® 3000扫描仪(型号:00-00212,购自美国Affymetrix公司)进行扫描,用Command Console 4.0软件(购自美国Affymetrix公司)读取原始数据,质控合格的数据采用R软件中Affy包进行归一化处理,所用的算法为MAS 5.0。
1.2.5 芯片数据生物信息学分析
采用GO富集分析、KEGG通路、STING在线分析工具等生物信息学方法分析前列腺癌及前列腺炎症组织中差异表达基因,并通过对重要节点基因文献挖掘的方法,找到与前列腺癌的发生、发展相关的基因。
2 结 果
2.1 芯片检测质量判断
2.1.1 RNA质检结果
所抽提的总RNA经超微量分光光度计NanoDrop检测A260nm和A280nm值,算得A260nm/ A280nm均大于1.8,表明所提取总RNA纯度较高;经Agilent Bioanalyzer 2100定量电泳仪检测,RIN(RNA完整系数)大于7.0且28s/18s大于0.7 (图1)。符合电泳质控标准,表明总RNA质量完好,可以进行芯片实验(表1)。
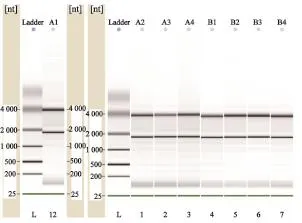
图 1 8例样本的电泳图Fig. 1 Electrophoresis map of 8 samples
2.1.2 基因表达谱芯片荧光信号扫描结果
采用GeneChip® Scanner 3000基因芯片扫描系统对8个样本芯片进行扫描,其荧光信号扫描图显示:芯片信号整体分布均匀,阳性质控探针信号清晰,阴性质控探针无明显信号存在,可以进行后续数据分析。
2.2 基因芯片检测结果的分布情况
2.2.1 芯片数据的散点图分析
芯片的原始数据经过标准化处理,转化为log2的值后,在一个二维坐标系中绘制散点图。分布在对角线两侧平行线上的点是表达水平无明显差异的基因,分布在对角线之外的点为表达有差异的基因,且距离对角线越远表示表达差异倍数越大,分布在对角线两侧平行线之外的点为差异表达倍数大于2倍的基因。如图2所示,表达谱芯片的散点图中的点分布大部分集中在对角线两侧平行线之外,说明存在许多表达显著改变的基因。
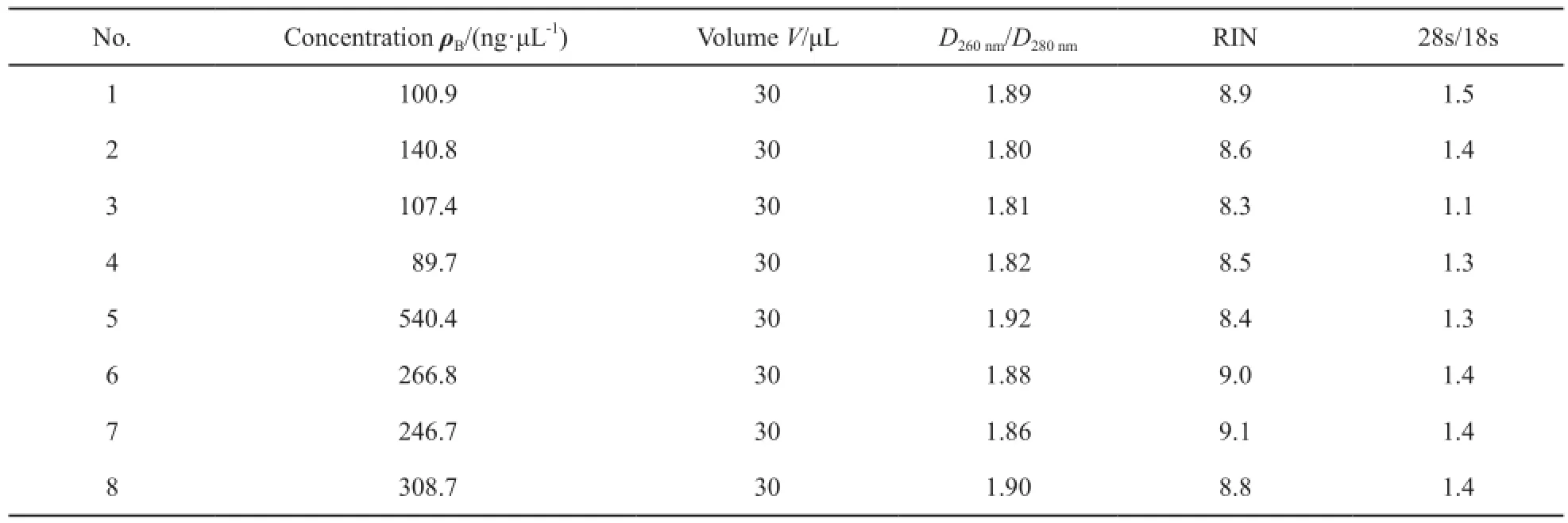
表 1 总RNA的质量检测结果Tab. 1 The results of total RNA detection
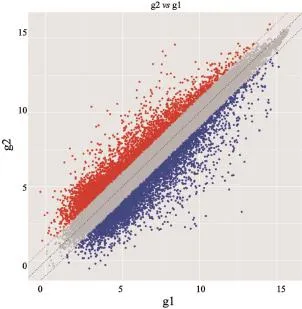
图 2 R软件归一化处理样本原始数据的散点图Fig. 2 The scatter plot of the normalized raw data with R software
2.2.1 芯片数据的热图分析
热图直观地显示了各个样本中不同基因的表达水平。黑色代表0,表示基因表达水平没有变化,红色代表表达水平升高,绿色代表表达水平降低;颜色的亮度代表基因表达水平升高或降低的程度。表达相似的基因(探针)和样本聚类在一起。如图3所示,可以看出炎性组织和前列腺癌组织间的表达谱明显不同。
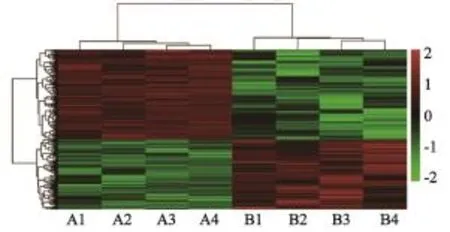
图 3 R软件归一化处理样本原始数据的热图Fig. 3 The heatmap of the normalized raw data with R software
2.3 基因芯片检测的数据结果及生物信息学分析
2.3.1 基因表达谱芯片数据结果
本研究选择4例确诊的恶性程度较高的前列腺癌组织标本,以4例前列腺炎症组织作为对照,按照Fold Change大于等于2,t检验P<0.05的筛选条件,筛选与前列腺癌有关的基因。结果发现表达差异基因1 819条中上调的有1 025条,下调的有794条。以Fold Change大于等于5、t检验P<0.01为条件进一步筛选基因,结果发现表达差异基因241条,其中上调的有128条,下调的有113条,部分上调和下调基因见表2,其中负号表示与炎症组织比较,在癌组织中该基因表达下调。
2.3.2 差异表达基因的相互作用网络分析
通过STING在线工具对241个前列腺癌差异基因的蛋白-蛋白相互作用网络进行分析。结果显示,在整个网络中,TPX2、ANLN、
NUSAP1、MELK、DLGAP5、KIF11、TOP2A、CDC6、RRM2、PBK、ASPM、KIF20A、CCNB1、PBC1、CDK1、BUB1、CDC20、NCAPG、NUF2、CENPF、CEP55、 CENPA、UBE2C、MUC、EZH2、CCN、CKAP2和MLF1IP6蛋白与其他大于等于5个蛋白存在相互作用关系,这些蛋白节点为蛋白互作网络的中心节点(图4)。
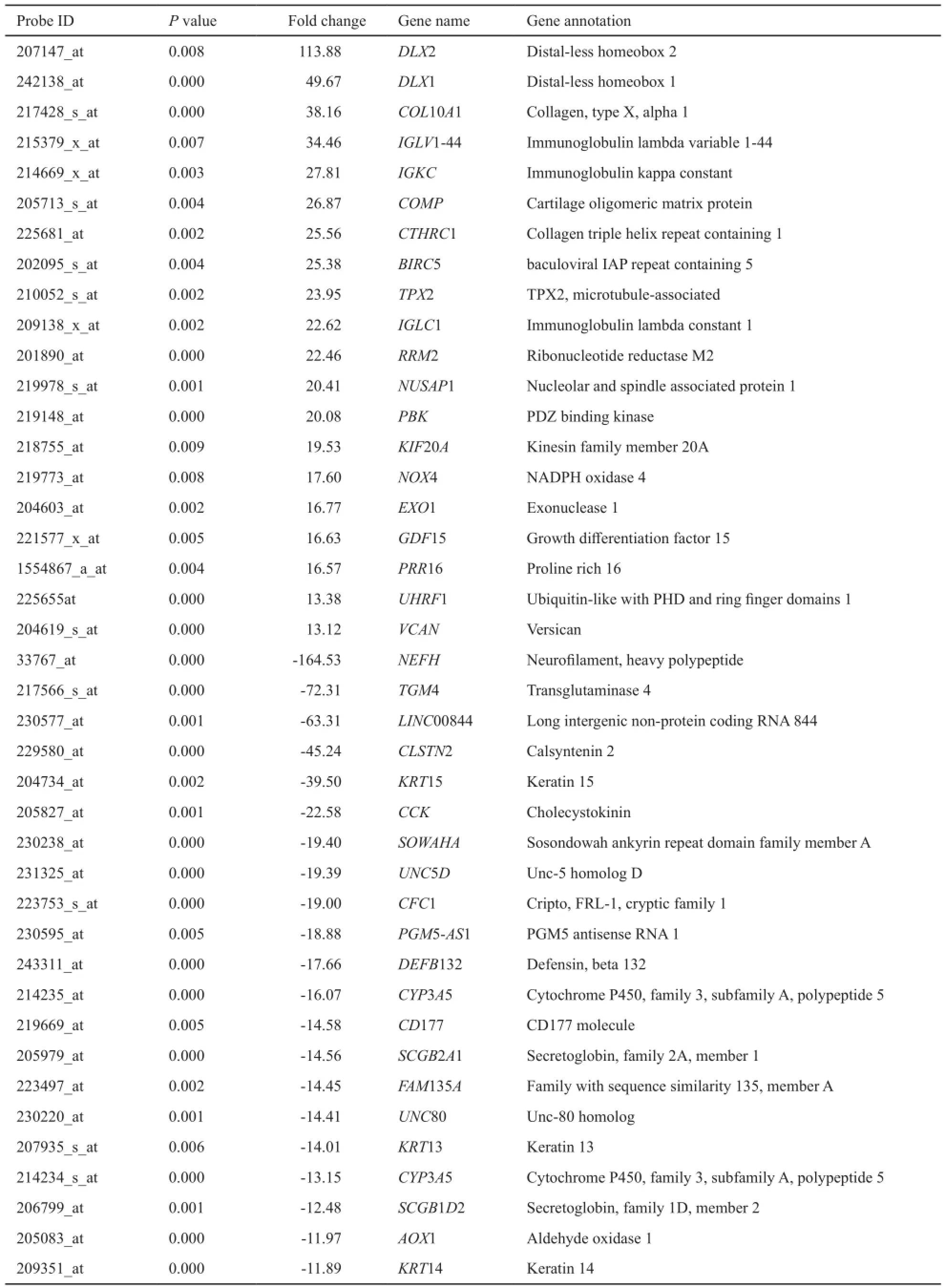
表 2 部分上调和下调的差异表达基因(癌/炎症组织)Tab. 2 Parts of up-regulated and down-regulated differentially expressed genes (cancer/inflammation)
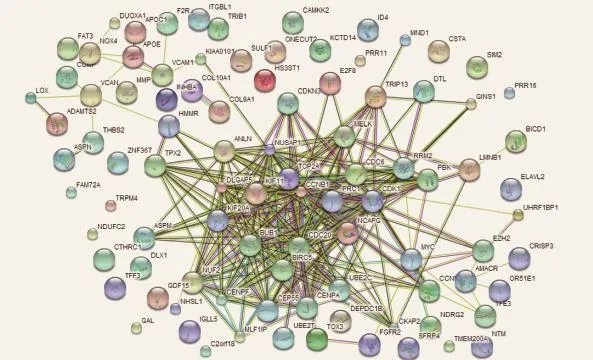
图 4 差异表达基因的蛋白与蛋白相互作用图Fig. 4 The protein-protein interactions among differentially expressed genes
2.3.3 差异表达基因的GO富集及KEGG通路分析
通过GO富集分析发现,上述差异基因主要涉及上皮细胞的生长、细胞周期的进程、细胞移行、纤维母细胞生长因子受体信号通路调节、细胞骨架蛋白结合小分子代谢过程、细胞表面受体信号通路、细胞黏附、细胞调节氨基酸代谢过程等分子功能、细胞组成以及生物学过程。
同时将基因芯片差异表达基因作KEGG通路分析,发现这些基因显著富集在谷胱甘肽代谢、p53信号通路、细胞周期、细胞外基质受体相互作用、蛋白的消化和吸收以及 TGF-β信号通路上(表3)。
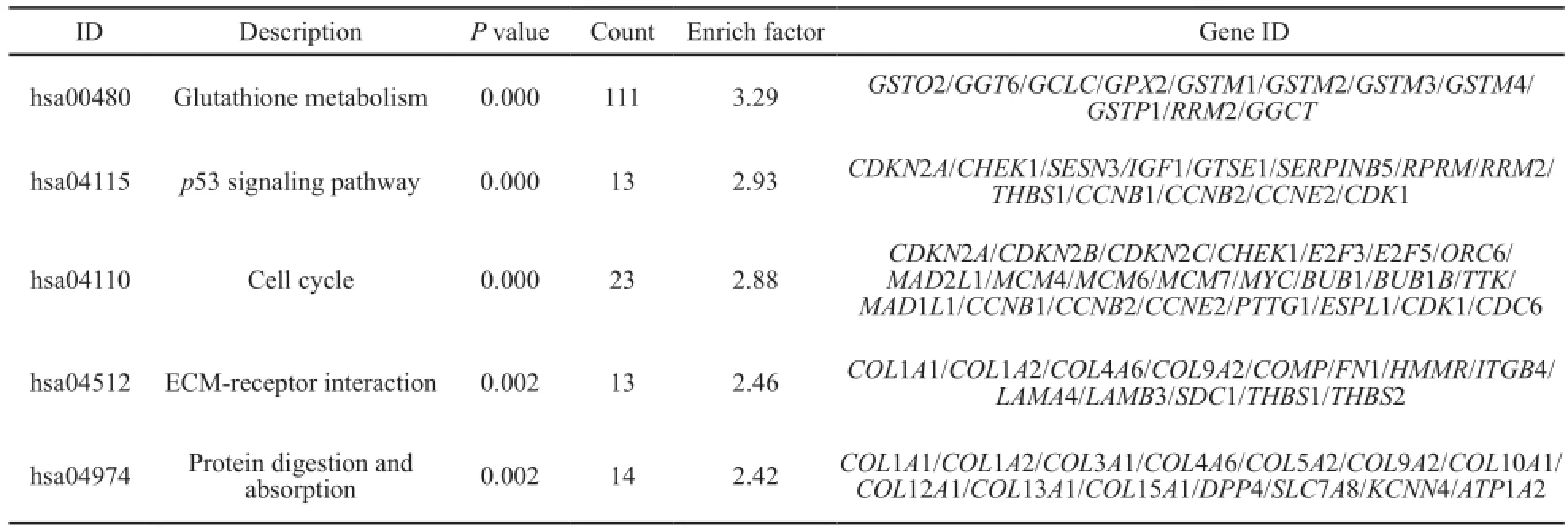
表 3 差异表达基因KEGG通路路径列表Tab. 3 The KEGG pathways of differentially expressed genes
3 讨 论
前列腺癌的发生是一个复杂的过程,是许多肿瘤相关基因表达失常或多种肿瘤抑制基因失活所致,因此在肿瘤组织癌变过程中对相关基因及基因组的变化研究显得尤为重要[1,5]。随着人类基因组计划的完成,基因芯片作为后基因组时代发展起来的一项DNA分析技术,具有快速、高效、平行化地检测大规模基因表达的优势,结合基因芯片表达数据的生物信息学分析,为我们更好地理解前列腺癌发生、发展的机制提供了有益的线索。本实验中我们采用了美国Affymetrix公司的U133 plus 2.0寡聚核苷酸点阵基因表达谱芯片,在前列腺癌组织中筛选出1 819条差异表达的前列腺癌相关基因,表明前列腺癌的发生、发展是一个多基因、多途径相互作用和相互影响的复杂过程。
从基因的功能分类来看,这些差异表达的基因主要涉及上皮细胞的生长、细胞周期的进程、细胞移行、纤维母细胞生长因子受体信号通路调节、细胞骨架蛋白结合小分子代谢过程、细胞表面受体信号通路、细胞黏附、细胞调节氨基酸代谢过程等分子功能、细胞组成等生物学过程。通过对这些基因分类有助于我们从分子水平了解前列腺癌发生的机制,为寻找新的前列腺癌标志物和探索基因治疗提供依据。在KEGG通路分析中我们注意到前列腺癌组织中大量与代谢相关的基因表达,涉及糖原降解、胆固醇合成、脂肪酸的氧化、3-磷酸肌醇的生物合成、嘌呤核苷酸的合成、谷胱甘肽的代谢通路,表明在前列腺癌中脂类代谢、核酸代谢以及糖代谢都发生了显著改变,也说明了与前列腺炎症组织相比,前列腺癌细胞获取能量的范围更广,可以从碳水化合物到脂肪酸和氨基酸。这与多项研究报道的结果一致[6-9]。由于前列腺特异抗原(prostate specific antigen,PSA)在临床前列腺癌诊断中存在很多缺陷,因此,通过对代谢分子及相关基因的研究来找到新的特异度和灵敏度更高的肿瘤标志物,成为前列腺癌研究的一个新的热点。
对这些差异基因进行相互作用网络分析发现以TPIP13、TPX2、ANLN、NUSAP1、MELK、DLGAP5、KIF11、TOP2A、CDC6、RRM2、PBK、ASPM、KIF20A、CCNB1、PBC1、CDK1、BUB1、CDC20、NCAPG、NUF2、CENPF、CEP55、CENPA、UBE2C、MUC、EZH2、CCN、CKAP2、MLF1IP等基因为中心构成了相互作用的网络,其中TOP2A、CDC6、CCNB1、CDK1、BUB1、CDC20、UBE2C、MUC、EZH2和CCN等基因与前列腺癌的关系已有大量文献报道。我们通过文献挖掘发现目前CEP55和ANLN基因与前列腺癌的关系报道较少,且这两个基因可能与前列腺癌的发生和预后关系密切。CEP55蛋白是新近发现的典型卷曲螺旋蛋白家族成员之一,定位于人染色体10q23.33,其转录的cDNA由464个氨基酸构成,包含9个外显子[10]。其主要功能是锚定微管聚合相关蛋白,参与纺锤体形成,进而调控细胞增殖。既往研究显示,CEP55可能是通过PLKl和TEXl4等信号通路参与肿瘤多种生物学功能[11-12]。CEP55在前列腺癌中的报道很少,Shiraishi等[13]研究发现,在根治性前列腺癌切除术的术后复发患者中,CEP55表达量显著增高,且术后随访资料显示CEP55水平越高,患者生存率越低,提示CEP55与前列腺癌的预后密切相关。本实验中,CEP55表达上调5.5倍,也提示在前列腺癌中该基因可能存在过表达现象。但其在前列腺癌发生、发展过程中的作用以及具体机制还不明确,值得进一步研究。ANLN是一种肌动蛋白结合蛋白,位于染色体7p14.2,全长84 kb,编码1 124个氨基酸。ANLN作为卵裂沟形成的一个关键蛋白,在细胞有些分裂过程中发挥重要作用。研究发现,ANLN在多种肿瘤中存在过表达现象[14-16]。Zhou等[17]研究发现,ANLN在乳腺癌组织中存在过表达现象,进一步在乳腺癌细胞中敲除ANLN基因发现,细胞生长受到抑制,并且可通过显著提高Cdc2的磷酸化水平和降低Cyclin D1的表达来抑制乳腺癌细胞的转移。同时发现,在敲除ANLN基因后大多数乳腺癌细胞处于G2/M期,这也与ANLN基因参与调节细胞质分裂的功能一致。ANLN蛋白在前列腺癌中的研究报道甚少,Tamura等[18]等在激素抵抗性前列腺癌组织标本的全基因组表达谱中发现ANLN存在过表达现象,与本实验结果一致。
综上所述,前列腺癌的发生、发展涉及到许多基因的异常表达,而这些异常表达的基因参与了多种分子生物学过程,深入研究这些基因将有利于进一步了解前列腺癌的发病机制。GO和KEGG通路分析结果也说明了前列腺癌的发生、发展是一个多因素的过程,虽然涉及到许多基因,但其中相当一部分可能是继发性改变,所以只有进一步筛选出关键的基因或信号通路,才有可能为疾病的诊断、治疗提供有效的指导,为建立个体化治疗方案和预后评估提供帮助。
[1] PASTUSZAK A W, PEARLMAN A M, GODOY G, et al. Testosterone replacement therapy in the setting of prostate cancer treated with radiation[J]. Int J Impot Res, 2012, 25(1): 24-28.
[2] MENGUAL L, ARS E, LOZANO J J, et al. Gene expression profiles in prostate cancer: identification of candidate noninvasive diagnostic markers[J]. Actas Urol Esp, 2014, 38(3): 143-149.
[3] 王建明, 史本康, 杨典东, 等. 应用基因芯片筛选前列腺癌差异表达基因的研究[J]. 中华实验外科杂志, 2012, 29(9): 1810-1812.
[4] GLINSKY G V, GLINSKII A B, STEPHENSON A J, et al. Gene expression profiling predicts clinical outcome of prostate cancer[J]. J Clin Invest, 2004, 113(6): 913-923.
[5] 石 玮, 董 莉. 前列腺癌相关分子研究进展[J]. 中华男科学杂志, 2015, 21(4): 357-362.
[6] LUCARELLI G, RUTIGLIANO M, GALLEGGIANTE V, et al. Metabolomic profiling for the identification of novel diagnostic markers in prostate cancer[J]. Expert Rev Mol Diagn, 2015, 15(9): 1211-1224.
[7] KAUSHIK A K, VAREED S K, BASU S, et al. Metabolomic profiling identifies biochemical pathways associated with castration-resistant prostate cancer[J]. J Proteome Res, 2014, 13(2):1088-1100.
[8] MELLER S, MEYER H A, BETHAN B, et al. Integration of tissue metabolomics, transcriptomics and immunohistochemistry reveals ERG- and gleason scorespecific metabolomic alterations in prostate cancer[J]. Oncotarget, 2016, 7(2): 1421-1438.
[9] ROBERTS M J, SCHIRRA H J, LAVIN M F, et al. Metabolomics: a novel approach to early and noninvasive prostate cancer detection[J]. Korean J Urol, 2011, 52(2): 79-89.
[10] JEFFERY J, SINHA D, SRIHARI S, et al. Beyond cytokinesis: the emerging roles of CEP55 in tumorigenesis[J]. Oncogene, 2016, 35(6): 683-690.
[11] BASTOS R N, BARR F A. Plk1 negatively regulates Cep55 recruitment to the midbody to ensure orderly abscission[J]. J Cell Biol, 2010, 191(4): 751-760.
[12] IWAMORI T, IWAMORI N, MA L, et al. TEX14 interacts with CEP55 to block cell abscission[J]. Mol Cell Biol, 2010, 30(9): 2280-2292.
[13] SHIRAISHI T, TERADA N, ZENG Y, et al. Cancer/testis antigens as potential predictors of biochemical recurrence of prostate cancer following radical prostatectomy[J]. J Transl Med, 2011, 9(1):153-162.
[14] OLAKOWSKI M, TYSZKIEWICZ T, JARZAB M, et al. NBL1 and anillin (ANLN) genes over-expression in pancreatic carcinoma[J]. Folia Histochem Cytobiol, 2009, 47(2): 249-255.
[15] PANDI N S, MANIMUTHU M, HARUNIPRIYA P, et al. In silico analysis of expression pattern of a Wnt/β-catenin responsive gene ANLN in gastric cancer[J]. Gene, 2014, 545(1): 23-29.
[16] AKINO Y, TESHIMA T, KIHARA A, et al. Carbon-ion beam irradiation effectively suppresses migration and invasion of human non-small cell lung cancer cells[J]. Int J Radiat Oncol Biol Phys, 2009, 75(2): 475-481.
[17] ZHOU W, WANG Z, SHEN N, et al. Knockdown of ANLN by lentivirus inhibits cell growth and migration in human breast cancer[J]. Mol Cell Biochem, 2015, 398(1-2): 11-19.
[18] TAMURA K, FURIHATA M, TSUNODA T, et al. Molecular features of hormone-refractory prostate cancer cells by genome-wide gene expression profiles[J]. Cancer Res, 2007, 67(11): 5117-5125.
Screening for differential genes of the prostate cancer and bioinformatics analysis of their interaction
XIA Qianlin1, SHAN Menglin1, DING Tao2, ZHU Yanjun3, HOU Jun4, ZHENG Jianghua1,5(1.Department of Laboratory Medicine, Shanghai Public Health Clinical Center, Fudan University, Shanghai 201508, China; 2. Department of Urology, Southern Branch of the Sixth People’s Hospital, Shanghai Jiao Tong University, Shanghai 201499, China; 3. Department of Urology, Zhongshan Hospital, Fudan University, Shanghai 200032, China; 4. Department of Pathology, Zhongshan Hospital, Fudan University, Shanghai 200032, China; 5. Department of Infection Control, Shanghai Public Health Clinical Center, Fudan University, Shanghai 201508, China)
ZHENG Jianghua E-mail: zhengjianghua2015@163.com
Background and purpose: Gene chip is a nucleic acid sequence analysis method which is based on hybridization. It is a high-through put assay which can widely detect the level of gene expression in different tissuesand cell types. This study aimed to compare and bioinformatically analyze differentially expressed genes between higher malignant degree of prostate cancer tissues and prostate inflammation tissues. Methods: The total RNAs were isolated from tissues of prostate cancer and prostate inflammation by TRIzol method and then purified, reversely transcribed to cDNA with incorporating biotin labeling probe, hybridized with Affymetrix Human U133 Plus 2.0 (covering 47 000 transcripts,representing 38 500 distinct genes). Picture signals of fluorescence in gene array were scanned and differential expression of gene in two tissues were compared by Command Console Software 4.0. These differential expressed genes were analyzed by bioinformatics methods finally. Results: According to the fold change ≥2, P<0.05, 1 819 differential expression genes including 1 025 up-regulated genes and 794 down-regulated genes were discovered. GO enrichment analysis displayed that these differentially expressed genes were mainly involved in cell cycle, cell metabolism, etc. KEGG pathway analysis found that these genes were mainly involved in some metabolism pathways including purine nucleotide metabolism. The interactions between the proteins encoded by these genes were analyzed by STING. Twenty key nodes genes including TPX2, ANLN, NUSAP1, MELK, DLGAP5, KIF11, TOP2A, RRM2 were discovered. Then this study revealed CEP55 and ANLN might be related to the occurrence and metastasis of prostate cancer by looking through literature. Conclusion: During the development of prostate cancer, the activation of genes related to cell cycle and cell migration, the abnormalities of genes related to metabolism and the inhibition of genes related to cell adhesion play critical roles in the development of prostate cancer. CEP55 and ANLN were related to the occurrence and prognosis of prostate cancer by systematic analysis which provided a valuable clue for the next experiment.
Prostate cancer;Gene chip;Differential genes;Bioinformatics
10.19401/j.cnki.1007-3639.2017.03.002
R737.25
A
1007-3639(2017)03-0169-08
2016-08-05
2016-12-30)
国家自然科学基金面上项目(81372318);上海市2014年度“科技创新行动计划”实验动物研究领域科技支撑项目重点项目(14140901400);上海市自然科学基金项目(13ZR1435000 )。
郑江花 E-mail:zhengjianghua2015@163.com
猜你喜欢
基础医学与临床(2022年1期)2022-01-21
今日农业(2021年4期)2021-06-09
天津医科大学学报(2021年2期)2021-03-29
知识文库(2020年5期)2020-05-11
心电与循环(2020年1期)2020-02-27
天津医科大学学报(2019年3期)2019-08-13
中国生殖健康(2019年7期)2019-01-06
江苏农业科学(2017年5期)2017-04-15
中国实用医药(2016年1期)2016-01-11
中国当代医药(2014年27期)2014-10-23
