
特应性皮炎的发病机制
2017-04-11 07:08罗金成宋志强
中华临床免疫和变态反应杂志 2017年4期
罗金成,宋志强
(第三军医大学西南医院皮肤科, 重庆 400038)
ChinJAllergyClinImmunol,2017,11(4):375- 381
特应性皮炎(atopic dermatitis, AD),又称特应性湿疹,是一种常见的以反复发作、皮损多形性、皮肤干燥和剧烈瘙痒等为基本临床特征的慢性炎症性皮肤疾病,其确切病因及发病机制尚未完全阐明,可能与遗传所致免疫失常、皮肤屏障功能障碍、环境因素及其交互作用密切相关[1]。有1/3的AD患儿病情反复迁延至成年,且常伴发哮喘及过敏性鼻炎等变态性疾病。特应性皮炎的发病率在过去几十年中显著增加,特别是在西方工业化国家,已经成为一个重要的健康问题[2]。
特应性皮炎的遗传学
AD具有明显的家族遗传倾向,很多AD患者存在与过敏性疾病相关的阳性家族史。Thomsen等[3]2007年对同卵孪生子的调查研究显示,决定AD发病的危险因素中,遗传因素占82%,而环境因素仅为18%。在同卵双生子中,AD的同时患病率约为80%,但在异卵双生子中却只为20%[4]。若父母中一方患AD,其后代患AD的发生率是59%;若父母均患AD,那么该比率将上升至81%[5]。以往的研究还发现,母亲患AD比父亲患此病的遗传危险性更高。这些流行病学调查均提示遗传因素在AD的发病中发挥重要作用。现已证实,AD的遗传方式不遵循孟德尔遗传定律,是多致病基因与环境因素共同作用所致的复杂疾病。
与AD发病相关的易感基因,主要包括皮肤屏障、固有免疫反应及特异性免疫反应相关基因3类。它们往往具有编码调控角质形成细胞终末分化相关蛋白以及固有或适应性免疫反应相关细胞因子的功能。以往的研究发现,一些皮肤屏障相关基因,诸如丝聚蛋白(filaggrin,FLG)基因、兜甲蛋白(loricrin,LOR)基因、外皮蛋白(involucrin,IVL)基因、表皮蛋白酶抑制剂相关基因(如SPINK5基因)及跨膜蛋白79(transmenbrane protein 7,TMEM79)等基因的功能突变、多态性和异常表达,往往与AD的发病密切相关[6]。固有免疫是机体抵御微生物侵袭的重要防线,其关键组分的基因,如核苷酸结合寡聚化结构域蛋白(nucleotide-binding oligomerization domain,NOD)1、NOD2、Toll样受体2(toll-like receptors 2,TLR2)、CD14以及防御素β1(defensin beta 1,DEFB1)等编码基因失常与AD发病相关。Th2型特异性免疫反应在AD的急性期起主导作用,与AD发病密切相关的Th2型细胞因子及趋化因子相关基因包括编码白细胞介素(interleukin,IL)- 4、IL- 5、IL- 6、IL- 9、IL- 13、胸腺基质淋巴细胞生成素(thymic stromal lymphopoietin,TSLP)、CC家族趋化因子受体5(CC chemokine receptor,CCR5)和FCER1A基因等。现已证实与AD发病相关的Th1型免疫反应基因为IL- 12、IL- 18基因等,在AD慢性期中扮演重要角色。AD的发病还与某些非Th1非Th2免疫途径相关基因发生突变有关,包括IL- 17、IL- 22、IL- 31等基因。
目前, FLG基因是文献报道最多、并被证实与AD发病高度关联的易感基因。基于对不同地区人群普查,发现10%~50%的AD患者FLG基因发生失活突变[7]。较存在杂合子FLG突变的AD患者而言,纯合子FLG突变者具有早年发病、掌纹症、变应原高致敏性以及其角质层pH值升高等临床特征。不过,只有10%左右的美国AD患者携带FLG突变基因,提示在AD的表型形成过程中发生主导作用的是其他危险因素。
AD具有明显的家族遗传背景,遗传因素在AD的发病中起重要作用。AD的遗传方式不遵循孟德尔遗传定律,是多致病基因与环境因素共同作用所致的复杂的遗传异质性疾病。与AD发病相关的易感基因,主要包括皮肤屏障、固有免疫反应及特异性免疫反应相关基因3类。近年来FLG基因已成为AD发病机制的研究热点。
特应性皮炎卫生学假说
2012年美国成人AD患病率为7%~10%,而在非工业化国家以及农村地区甚至更低[8]。针对这一流行病学现象,Strachan早在1989年就提出了特应性疾病的卫生学假说(hygiene hypothesis)。该假说认为:儿童早期暴露于存在诸多不同病原体(如寄生虫、病毒、细菌等)的环境中,可促使其尚未成熟的免疫系统不向Th2反应方向偏移,从而有助于防止过敏性疾患的产生[9]。以往的流行病学调查提示,早期接受过大量广谱抗生素和疫苗治疗的儿童,其患过敏性疾病的危险性显著增加。另据国外报道,儿童花粉热及湿疹的发病率与个体家庭中年长儿童的人数呈显著负相关性[10]。原因可能是,过去50年以来西方发达国家生育率的降低,导致与年长儿童的不良卫生接触所引起的年幼儿童感染发生率随之减少,而感染是预防AD发病的保护因素,从而使AD的发病率急剧上升。Ernst等[2]指出,在西方工业化国家中,与本国出生的儿童和青少年相比,有移民背景的儿童和青少年更容易罹患感染,也许是与后者的家庭人口规模较大,生活方式及生活环境的差异而使其更容易暴露于易发生感染的环境有关。个体家庭人口规模的减少及卫生条件的改善在极大降低儿童感染发生率的同时,也消除了感染对防止过敏症发生的保护作用。在卫生情况欠佳的发展中国家,儿童暴露于充满污垢及病原体的环境中,可能实际上起到防止AD等过敏性疾患发生的作用。这种卫生暴露或许可以解释在过去50年AD发病率上升及发病存在区域差异的原因。
卫生学假说认为,儿童早期持续暴露在过于卫生的生活环境,儿童早期接触微生物抗原缺乏,导致免疫系统缺乏感染的刺激而发育异常,从而使机体患过敏性疾病的风险性上升。Th1功能发育障碍,Th2功能相对强势是经典的卫生学假说的免疫学致病基础,但是单纯的Th1/Th2功能失衡发病机制并不能完全解释卫生学假说,例如在西方工业化国家,Th2型过敏性疾病发病率的升高的同时并不伴随Th1型自身免疫性疾病发病率的减少。但是这一假说无疑相对合理地解释了过去50年来西方工业化国家AD发生率大幅度增加的原因。
特应性皮炎皮肤屏障功能失常机制:outside-in病因假说
1999年Elias等[11]提出 outside-in假说,该假说认为皮肤屏障受损早在AD发病前就存在了,是导致机体免疫失常的启动因素。表皮结构蛋白相关基因缺陷、表皮结构蛋白合成减少或结构功能改变、过敏原刺激、搔抓等导致的物理损伤及微生物定植引起的化学生物性损伤等致病因素,均可致表皮屏障受损,进而引起机体免疫功能失常,最终导致全身性过敏性炎症的发生(图1)。这就是AD发病的遗传及获得性皮肤屏障功能失常假说。
皮肤屏障正常功能的发挥有赖于丝聚蛋白的正常表达。丝聚蛋白基因(FLG基因)缺陷是致皮肤屏障功能缺陷的重要因素。早在2006年McLean等对北欧人群进行追踪调查研究,发现FLG基因的失活突变与近20%的AD病例有关[12]。
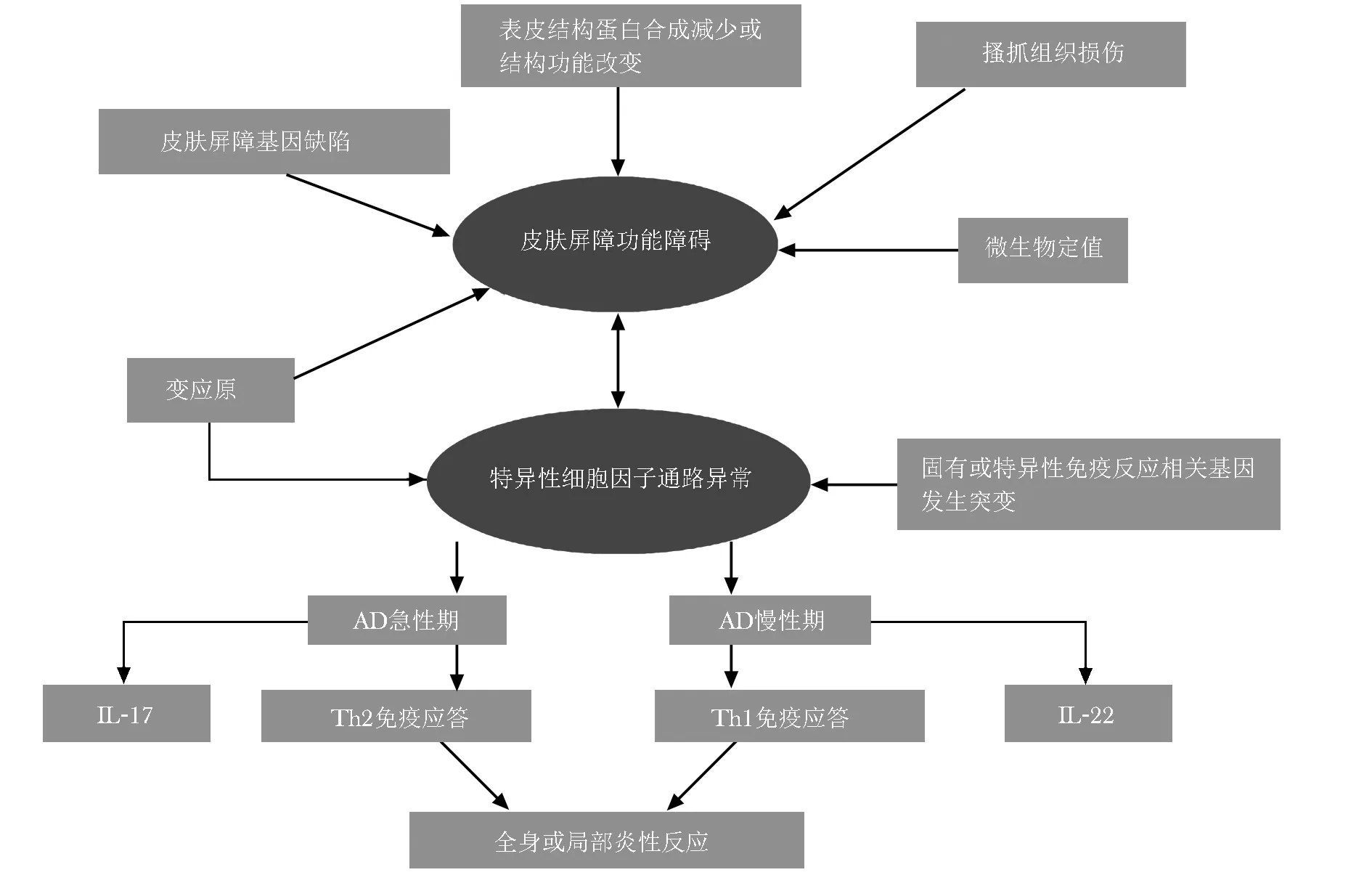
图1特应性皮炎发病机制示意图
Fig1Schematic diagram of pathogenesis of atopic dermatitis
AD:特应性皮炎; IL- 17:白细胞介素17; IL- 22:白细胞介素22
Palmer等[13]进行的相关研究表明,发生失活突变的丝聚蛋白基因与常染色体半显性遗传模式有关。丝聚蛋白的编码基因位于染色体1q21.3上,在体内被加工后最终分解生成大量的天然保湿因子(natural moisturizing factors,MNFs),在维持皮肤屏障功能上扮演重要角色。皮肤屏障功能缺陷不但可使机体易于遭受来自外界损伤因素的入侵并引起经表皮水分丢失(transepidermal water loss,TEWL)增加,而且TEWL水平与AD病情严重程度有关[14]。导致FLG突变及其在细胞水平上的致病机制至今尚未明了,但是已有研究发现FLG基因的失活突变可诱使表皮角质形成细胞形态上发生改变进而影响其正常功能,同时引起细胞外板层状体组织结构发生紊乱,从而导致皮肤屏障受损[15]。
FLG基因缺陷与AD的发病相关。FLG突变携带者不仅容易诱发顽固性、早年发病型AD,而且更容易伴发哮喘,食物过敏以及皮肤感染等等病症[16]。然而大部分AD患者FLG基因并未发生突变,即使FLG基因发生突变的大多数患者病情最终也得到缓解。因此单纯的FLG基因突变并不能解释所有AD发病机制。Suárez-Farias等[17]的研究显示,AD患者皮损区及非皮损区分布有多种终末分化蛋白,包括FLG、LOR、IVL、周斑蛋白、谷氨酰胺转移酶等,它们的表达受到显著抑制,其作用机制仍未清楚。
claudin蛋白是位于表皮颗粒层中角质形成细胞膜表面的一种紧密结合蛋白,它是表皮维持正常生理功能必不可少的结构,在角质层功能失常时可作为额外的屏障起作用。claudin蛋白与皮肤屏障损害与有着密不可分的关系。有文献报道,claudin- 1 蛋白基因缺乏可诱发致死性皮肤渗透性缺陷病。表皮中claudin蛋白合成减少或其正常结构功能发生改变,均可损伤角质层下的紧密连接正常功能,导致经表皮水分丢失增加,从而造成皮肤屏障受损。Kuo等[18]进行的研究已证实AD患者存在表皮claudin蛋白含量下降的现象。在AD患者中,表皮claudin蛋白的表达不仅与Th2标志物(血清总嗜酸粒细胞计数和总IgE水平)呈负相关,而且与皮损区Th2细胞因子水平呈负相关,这提示Th2细胞因子具有下调上皮细胞claudin蛋白的作用。
微生物定植在AD的发生和发展过程中发挥重要作用。持续定植于AD患者皮肤上的金黄色葡萄球菌可通过多种机制引发和加重AD皮肤炎症。具体作用机制如下:首先,金黄色葡萄球菌定植及其分泌的外毒素可作为超抗原激活相关炎症细胞,促进T细胞增殖及释放IL- 22、IL- 31等促炎症细胞因子参与炎症和组织损伤。其次,金黄色葡萄球菌可分泌大量的丝氨酸蛋白酶,加速表皮角质形成细胞过早脱落,导致角质层变薄,最终导致外界各种刺激物与过敏原的入侵。再者,正常皮肤表面的共生菌群可分泌抗微生物肽(antimicrobial peptides, AMPs),可抑制金黄色葡萄球菌过度繁殖;AD患者如常规应用局部抗生素可导致共生菌群受抑制而金黄色葡萄球菌却过度繁殖。最后,相对银屑病,AD患者皮肤中的抗微生物肽含量下降而金黄色葡萄球菌黏合分子表达增多。通过上述各种化学生物性致病机制,定植的金黄色葡萄球菌刺激表皮持续产生炎症反应,最终导致皮肤屏障功能受损。
上述诸多致病因素致表皮屏障受损后,机体皮肤的通透性上升,导致外源性变应原(如尘螨、花粉、金黄色葡萄球菌及其分泌的外毒素等)更易进入表皮,被皮肤树突状细胞(dendritic cells, DCs)识别、提呈并诱导随后的T细胞免疫反应,最终可导致系统性过敏性疾病的发生。Oyoshi等[19]的研究发现,皮肤屏障功能受损后,可诱发受损区表皮角质形成细胞释放胸腺基质淋巴细胞生成素(thymic stromal lymphopoietin,TSLP)增多。TSLP可激活树突状细胞(dendritic cells,DCs),后者可刺激Th0细胞向Th2细胞分化,从而促进Th2细胞的增殖并分泌Th2型细胞因子(诸如IL- 4、IL- 5、IL- 13等)和促炎症因子促使IgE、嗜酸粒细胞、嗜碱粒细胞、肥大细胞等的产生,进而引起过敏性炎症反应。
outside-in假说认为AD的发病是由皮肤向免疫、由外而内逐步发展的过程。outside代表着遗传因素或后天环境因素造成的皮肤屏障功能失常;in是指皮肤屏障缺陷后引起机体免疫失常进而产生全身性过敏性炎症[20]。目前该假说仍无法完全清楚阐明AD的自然病程的每个环节,比如表皮屏障受损后引起机体免疫功能失常的作用途径有哪,具体作用机制是什么等,想对其机制彻底的了解还需要进行更多的研究。
特应性皮炎免疫功能紊乱机制:inside-out发病假说
尽管近几年学术界对AD发病的遗传及获得性皮肤屏障功能失常假说比较推崇,但是目前却有越来越多的证据支持inside-out AD发病假说。该假说认为,在遗传因素(固有或特异性免疫反应相关基因缺陷)与环境因子(过敏原、搔抓、微生物定植等)的共同作用下,机体内部某些特异性细胞因子通路被激活,诱发全身过敏性炎症,进而致使角质形成细胞分化缺陷,最终导致皮肤屏障功能失常。免疫功能紊乱是否在AD的发生发展进程中起主导作用,尚存在较大争议。现已证实,AD的异常免疫应答涉及数个环节,如皮肤DCs及朗格汉斯细胞对变应原的识别加工与呈递、Th2占主导的异常免疫反应、调节性T细胞功能受损、IgE过度表达和嗜酸粒细胞增加等[21]。
AD的发病与许多固有或特异性免疫反应相关基因发生突变有关。传统上认为,AD发病具有双相免疫模式:AD急性发作期时以Th2反应为主,激活Th2细胞产生大量的IL- 4、IL- 5、IL- 6、IL- 9、IL- 10和IL- 13等Th2细胞因子,进而使B细胞过量释放IgE,嗜酸粒细胞计数增加,以介导过敏反应和体液免疫应答为主;在AD慢性期,则表现为以产生干扰素γ(interferon γ,IFN-γ)、肿瘤坏死因子β(tumor necrosis factor β,TNF-β)、IL- 2、IL- 12、IL- 18基因等Th1细胞因子为主的Th1细胞占优势的免疫应答。近些年来,有国外学者提出,Th2反应,在急性期和慢性期AD损伤中均主导作用;而Th1细胞介导的炎症反应,在慢性AD损伤中扮演重要角色[22]。
目前对AD急性期异常激活的IL- 4、IL- 9、IL- 17等细胞因子通路的研究较为热门。IL- 4是一种经典的Th2型细胞因子,可促使Th0细胞向Th2细胞分化,并诱导IgE类型转换。IL- 4通路的异常激活与AD发病存在明显的关联性。IL- 4可通过JAK1,3-STAT6信号传导通路诱导小鼠持续表达STAT6并最终致使该小鼠产生AD样皮损[8]。IL- 9可增强IL- 4促进Th2细胞分化的作用。Hong等[23]的研究显示急性AD患者皮肤中IL- 9和IL- 9受体表达明显升高,且外周血IL- 9水平与临床病情严重程度呈显著相关。IL- 17主要由Th17细胞产生,可启动协同多种前炎症细胞因子和趋化因子参与AD急性期的炎症反应。马蕾等[24]的研究发现急性期AD患者外周血中IL- 17 mRNA表达水平及血清中IL- 17含量亦明显升高。国外有报道,Th17/IL- 12/IL- 23通路参与AD的发病进程,但其具体作用机理尚未明确。
近期对AD慢性期细胞因子除了经典的Th1型细胞因子外,研究较多的是IL- 22。目前有关IL- 22通路的研究存在矛盾。慢性期AD皮损中存在Th22细胞浸润现象,皮损区Th22细胞数目与AD病情严重程度密切相关。AD皮损中的IL- 22主要由Th22细胞产生,能够通过下调角质形成细胞的分化基因的表达来抑制其分化,并促进角质形成细胞的增殖和表皮增生。而张丽明等[25]在文献中指出,IL- 22可通过STAT 信号传导通路下调包括FLG、LOR、IVL、周斑蛋白等多种表皮终末分化蛋白的表达,抑制FLG转录后加工酶的表达,加重表皮损伤。
临床上应用IL- 4、IL- 13、IL- 22等细胞因子于受试者后,可以观察到受试者表皮中的角质形成细胞终末分化蛋白出现抑制状态。近年在国外,一些针对特异性细胞因子通路的靶向免疫拮抗剂已开始走进临床。IL- 4受体拮抗剂(dupilumab),可阻断IL- 4及IL- 13通路的信号传导,在治疗中重度AD的临床试验中效果满意[26]。IL- 12拮抗剂(乌思奴单抗,ustekinumab)、抗IgE抗体(奥马珠单抗,omalizumab),已获批进入AD临床试验[27- 28]。这些报道在一定程度上从临床上反证了inside-out发病假说。
近10年来,通过对AD异常免疫应答机制研究的深入,许多特异性细胞因子通路的作用机制得以解晰,伴随而来的是特异性细胞因子通路拮抗剂的不断问世,引发了AD治疗学上的重大革命。inside-out发病假说日益受到人们的重视。
结 论
AD的发生是与皮肤屏障受损及免疫功能紊乱密切相关的复杂的多因素遗传异质性炎症性皮肤病。但皮肤屏障受损及免疫功能紊乱两者在AD发病过程中发生的先后顺序、由谁主导以及各自所起的精确作用机制尚不完全清楚,今后从遗传学及免疫学领域探讨这两个假说与AD关系将成为研究AD发病机制的热点,通过深入解析AD的发病机制将有助于研发更多有效治疗AD的方法。
[2]Ernst SA, Schmitz R, Thamm M, et al. Lower preva-lence of atopic dermatitis and allergic sensitization among children and adolescents with a two-sided migrant background[J]. Int J Environ Res Public Health, 2016, 13 pii: E265.
[3]Thomsen SF, Ulrik CS, Kyvik KO, et al. Importance of genetic factors in the etiology of atopic dermatitis: a twin study[J]. Allergy Asthma Proc, 2007, 28: 535- 539.
[4]Malajian D, Guttman-Yassky E. New pathogenic and therapeutic paradigms in atopic dermatitis[J]. Cytokine, 2015, 73: 311- 318.
[5]李丽莎, 尹佳. 特应性皮炎遗传学发病机制[J]. 中华临床免疫和变态反应杂志, 2015, 9:49- 54.
[7]Leung DY. Our evolving understanding of the functional role of filaggrin in atopic dermatitis[J]. J Allergy Clin Immunol, 2009, 124: 494- 495.
[8]Kantor R, Silverberg JI. Environmental risk factors and their role in the management of atopic dermatitis[J]. Expert Rev Clin Immunol, 2017, 13: 15- 26.
[9]Penders J, Gerhold K, Thijs C, et al. New insights into the hygiene hypothesis in allergic diseases: mediation of sibling and birth mode effects by the gut microbiota[J]. Gut Microbes, 2014, 5: 239- 244.
[10] Brüssow H. Turning the inside out: the microbiology of atopic dermatitis[J]. Environ Microbiol, 2016, 18: 2089- 2102.
[11] Elias PM, Wood LC, Feingold KR. Epidermal pathogenesis of inflammatory dermatoses[J]. See comment in PubMed Commons below Am J Contact Dermat, 1999, 10: 119- 126.
[12] Brown SJ, Irvine AD. Atopic eczema and the filaggrin story[J]. Semin Cutan Med Surg, 2008, 27: 128- 137.
[13] Palmer CN, Irvine AD, Terron-Kwiatkowski A, et al. Common loss-of-function variants of the epidermal barrier protein filaggrin are a major predisposing factor for atopic dermatitis[J]. Nat Genet, 2006, 38: 441- 446.
[14] Gupta J, Grube E, Ericksen MB, et al. Intrinsically defective skin barrier function in children with atopic dermatitis correlates with disease severity[J]. J Allergy Clin Immunol, 2008, 121: 725- 730.
[15] Elias PM, Hatano Y, Williams ML. Basis for the barrier abnormality in atopic dermatitis: outside-inside-outside pathogenic mechanisms[J]. J Allergy Clin Immunol, 2008, 121: 1337- 1343.
[16] Liang Y, Chang C, Lu Q. The Genetics and Epigenetics of Atopic Dermatitis-Filaggrin and Other Polymorphisms[J]. Clin Rev Allergy Immunol, 2016, 51: 315- 328.
[18] Kuo IH, Carpenter-Mendini A, Yoshida T, et al. Activation of epidermal toll-like receptor 2 enhances tight junction function: implications for atopic dermatitis and skin barrier repair[J]. J Invest Dermatol, 2013, 133: 988- 998.
[19] Oyoshi MK, Larson RP, Ziegler SF, et al. Mechanical injury polarizes skin dendritic cells to elicit a T(H)2 response by inducing cutaneous thymic stromal lymphopoietin expression[J]. J Allergy Clin Immunol, 2010, 126: 976- 984, 984.
[20] 瞿旻晔, 袁晓琳, 马健. 特应性皮炎发病过程及其机制研究现状探讨[J]. 中国皮肤性病学杂志, 2015, 29:1085- 1087.
[21] 中华医学会皮肤性病学分会免疫学组. 中国特应性皮炎诊疗指南(2014版)[J]. 中华皮肤科杂志, 2014,47:603- 606.
[22] Gittler JK, Shemer A, Surez-Farias M, et al. Progressive activation of T(H)2/T(H)22 cytokines and selective epidermal proteins characterizes acute and chronic atopic dermatitis[J]. J Allergy Clin Immunol, 2012, 130: 1344- 1354.
[23] Hong CH, Chang KL, Wang HJ, et al. IL- 9 induces IL- 8 production via STIM1 activation and ERK phosphorylation in epidermal keratinocytes: A plausible mechanism of IL- 9R in atopic dermatitis[J]. J Dermatol Sci, 2015, 78: 206- 214.
[24] 马蕾, 薛海波, 赵翠阳, 等. 特应性皮炎患者外周血Th17细胞比例及不同细胞因子微环境对其分化的影响[J/CD]. 中华临床医师杂志(电子版), 2012, 6: 5440- 5444.
[25] 张丽明, 段姚尧, 董小青, 等. Th22细胞及其效应因子IL- 22与炎症性皮肤病关系的研究进展[J]. 中国药理学通报, 2014, 30: 905- 907.
[26] Beck LA, Thaçi D, Hamilton JD, et al. Dupilumab treatment in adults with moderate-to-severe atopic dermatitis[J]. N Engl J Med, 2014, 371:130- 139.
[27] Puya R, Alvarez-López M, Velez A, et al. Treatment of severe refractory adult atopic dermatitis with ustekinumab[J]. Int J Dermatol,2012, 51: 115- 116.
[28] Saini S, Rosen KE, Hsieh HJ, et al. A randomized, placebo-controlled, dose-ranging study of single-dose omalizumab in patients with H1-antihistamine-refractory chronic idiopathic urticaria[J]. J Allergy Clin Immunol, 2011, 128: 567- 573.
猜你喜欢
江苏安全生产(2022年8期)2022-11-01
今日农业(2022年3期)2022-06-05
小资CHIC!ELEGANCE(2021年36期)2021-10-15
建材发展导向(2021年14期)2021-08-23
皮肤病与性病(2021年3期)2021-07-30
智慧健康(2021年33期)2021-03-16
昆明医科大学学报(2020年12期)2021-01-26
中华养生保健(2020年10期)2021-01-18
艺术评鉴(2020年5期)2020-04-30
祝您健康·文摘版(2019年4期)2019-06-11
