
团簇Mn3BP的电子自旋密度
2017-04-10 07:22方志刚崔远东徐诗浩李雯博
辽宁科技大学学报 2017年6期
周 帅,方志刚,崔远东,张 伟,徐诗浩,刘 琪,冯 天,李雯博
(辽宁科技大学 化学工程学院,辽宁 鞍山 114051)
原子簇是指由原子(或分子)结合在一起的团体结构,它是介于原子(或分子)与固体粒子之间的团粒分子。近几十年来由于金属原子簇化合物的催化功能[1-2]、生物金属原子簇[3-4]、超导及新型材料[5]等方面的研究需要,促使金属原子簇化学快速发展。金属原子簇具有较高的耐磨及抗腐蚀性[6]、良好的催化活性[7-8]、优异的磁学性能[9-12]及超导性能[13]等,使其在制备新型材料[14]、催化剂[15]、储氢材料[16]及超导材料[17-18]等领域得到广泛的应用。过渡金属元素与类金属元素硼组成的团簇是目前的研究热点,对Mn-B二元体系和含Mn、B、P多元体系研究仅有少量报道[19-22],对Mn-B-P三元体系的研究还未见相关报道。本文将对Mn-B-P三元体系的原子簇进行研究,以Mn3BP为模型,对团簇Mn3BP的电子自旋密度进行较深层次的研究,从而推断其相关性质,希望能给相关的研究做出一些有价值的参考。
1 计算方法
首先根据拓扑学原理和化学键理论[23-24],设计出了团簇Mn3BP二、四重态的所有可能构型。之后利用含相关校正的DFT[25]方法,在B3LYP/Lan12dz水平下对原子簇Mn3BP的所有可能构型进行全参数优化和频率计算,得到其稳定的构型。计算时对金属锰原子采用Hay[26]等人的含相对论校正的有效核电势价电子从头计算基组,即采用18-eECP双ξ基组(3s,3p,3d/2s,2p,2d),且P原子加加极化函数ξP.d=0.55[27];所有计算均在启天M7150微机上采用Gaussian09程序完成。
2 结果与讨论
2.1 稳定构型
采用上述模型和计算方法分别对团簇Mn3BP二、四重态构型进行优化计算,排除含有虚频的不稳定构型和几何构型相同的构型,最终得到四重态稳定构型5种,如图1中的1(4)~5(4)(右上角括号数字表示重态,下同),得到二重态稳定构型4种,如图1中1(2)~4(2),二、四重态构型分别按能量由低到高排序。图1中各构型中的数字表示各键键长(单位:nm),为方便研究,以能量最低的构型1(4)作为基准,令其能量为0 kJ/mol,然后依次得到其它构型的相对能量(括号里的数值表示相对能量)。
图1中可以看出,各稳定构型的几何构型有戴帽三角锥(1(4)和 4(4)),三角双锥(5(4)、1(2)和 2(2)),平面四边形(3(4)和3(2))和平面五边形(2(4)和4(2))。构型1(4)是以Mn1-Mn2-Mn3为基准面,B为锥顶,P为帽顶的戴帽三角锥;构型4(4)是以B-Mn1-Mn3为基准面,P为锥顶,Mn2为帽顶的戴帽三角锥。构型5(4)和 2(2)均是以 Mn1-Mn2-Mn3 为基准面,P 为锥顶,B为锥底的三角双锥,优化构型相同,多重度不同;构型1(2)是以Mn1-Mn3-B为基准面,P为锥顶,Mn2为锥底的三角双锥。构型 3(4)和 3(2)均是B原子作为中心原子,四边顶点以Mn1-Mn2-P-Mn3的排列顺序构成的平面四边形。构型2(4)和4(2)是由Mn1、Mn2、Mn3、B、P构成的平面五边形。
2.2 各构型的自旋密度分布
构型的稳定性有很多影响因素,电子自旋密度分布是其中的重要影响因素,分析各构型的自旋密度分布可以帮助分析各构型的稳定性。表1列出了各优化构型原子的电子自旋密度值,正值的含义是α成单电子出现的净概率密度,负值则代表β成单电子出现的净概率密度。分析表1中数据可知,所有构型的B原子的电子自旋密度均为负值,而P原子的电子自旋密度也均为负值(除了构型1(4)外),由此可知各构型中(除了构型1(4)外)的非金属原子B、P上的电荷分布都是自旋向下的β电子。从整体来看,所有构型中(除构型5(4)外)有一个Mn原子的电子自旋密度值为负数,其余两个Mn原子的电子自旋密度为正数,也就表示每个构型中(除构型5(4)外)有一个Mn原子的电荷分布是自旋向下的β电子,其余两个Mn原子的电荷分布是自旋向上的α电子,构型4(4)、5(4)、2(2)、3(2)有两个Mn原子的电荷分布是自旋向上的α电子,且电子自旋密度值相同或近似相等。
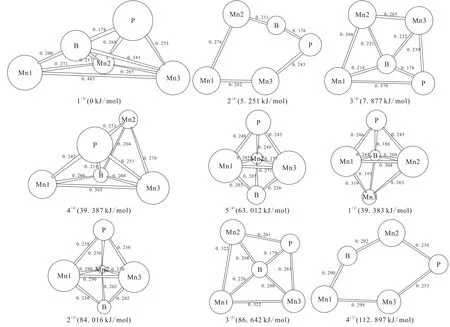
图1 团簇Mn3BP优化构型图Fig.1 Optimized configurations of cluster Mn3BP
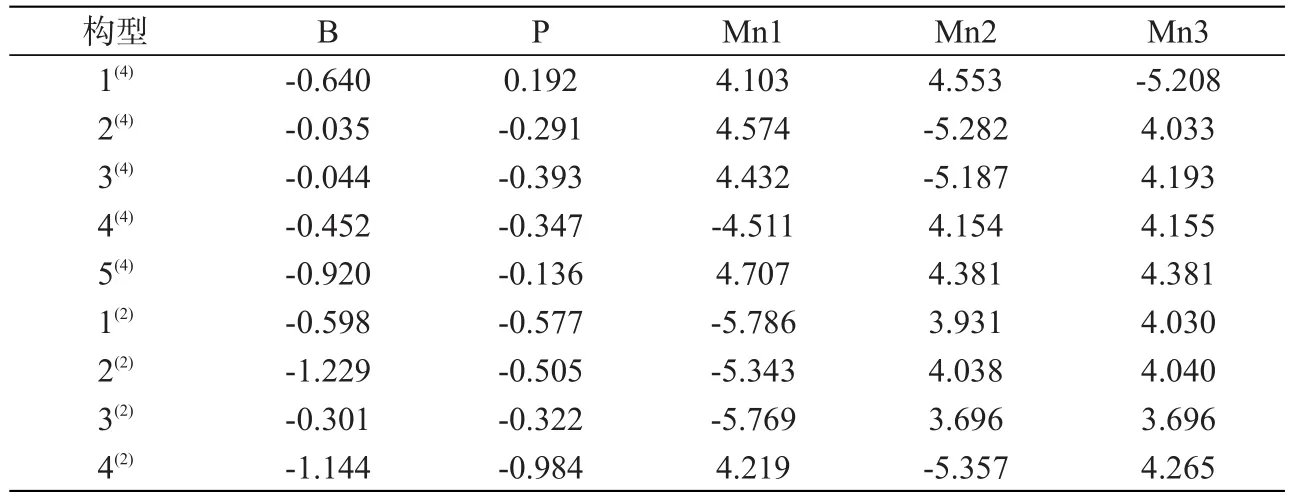
表1 团簇Mn3BP各构型原子的电子自旋密度值Tab.1 Spin density of each configuration atom in cluster Mn3BP
只从原子的电子自旋密度分析,存在很大的局限性,并不能得到很准确的结论。为了能更好地研究电子自旋密度对构型稳定性的影响,需要分析α电子、β电子及其重叠部分对其稳定性的影响。表2列出了团簇Mn3BP的各构型原子间的电子自旋密度值,正值代表原子间成键时α电子过剩,负值代表原子间成键时β电子过剩。为更方便地研究自旋密度分布对构型稳定性的影响,图2画出了团簇Mn3BP的各构型相对应的自旋密度分布图,按能量高低排序,图中浅灰色代表α电子,深灰色代表β电子。
依据表2的电子自旋密度值来分析内部原子间α、β电子分布情况,及其引起的构型各原子间的成键强弱和均匀度,从而影响各构型的稳定性。所有构型中非金属原子B与P之间的电子自旋密度值全都为正值,说明非金属原子间成键时α电子过剩;又通过表1知所有构型中(除构型1(4)外)的非金属原子B、P的电子分布为β电子;以上两点说明了B、P之间成键时的电子很大程度上受金属原子Mn的影响。结合表2数据和图2分析,构型1(4)与其他构型相比,构型原子间电子分布密度大且较为均匀,有效降低该构型的能量,使构型1(4)更加的稳定;构型4(2)的原子间电子分布密度小且不均匀,且对称性也不好,导致该构型的能量高,稳定性差。
为更好地寻找电子自旋密度分布对构型稳定影响的规律,将各构型的电子自旋密度分布图进行分类讨论,大致将其分为了三类。第一类:全部构型中最为稳定的前两种构型,包括构型1(4)和构型2(4)。这两种构型的电子自旋密度分布比较均匀,且具有对称性较好,构型1(4)的B-Mn原子间只有B-Mn1有β原子过剩,P-Mn原子间只有P-Mn2有β原子过剩,Mn-Mn原子间只有Mn1-Mn2有β原子过剩;构型2(4)的B-Mn原子间只有B-Mn3有α电子过剩,P-Mn原子间只有P-Mn2有α电子过剩,Mn-Mn原子间只有β电子过剩;构型 1(4)的 P-Mn2和P-Mn3之间的电子自旋密度值绝对值近似相等(P-Mn2:-0.095;P-Mn3:0.092),Mn1-Mn2和Mn2-Mn3之间的电子自旋密度值的绝对值也近似相等(Mn1-Mn2:-0.125;Mn2-Mn3:0.126);构型 2(4)的Mn1-Mn2和Mn2-Mn3的电子自旋密度值近似相等(Mn1-Mn2:-0.026;Mn2-Mn3:-0.029);以上几点说明了构型1(4)的原子间电子自旋密度分布比构型2(4)更均匀,原子间成键强度大且均匀,能量较低,比构型 2(4)的稳定性好。
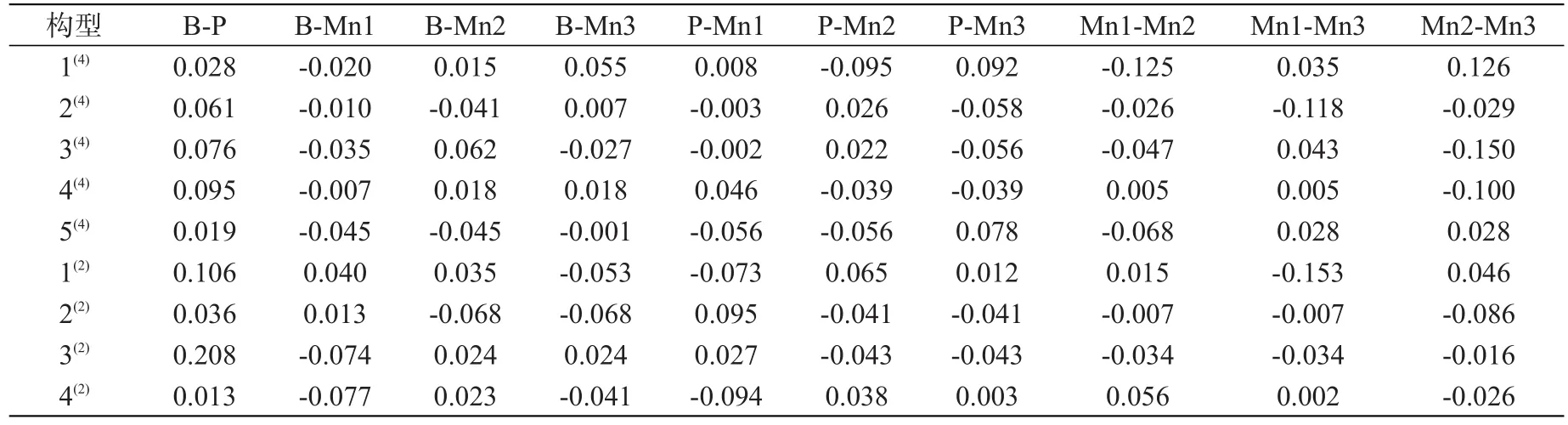
表2 团簇Mn3BP各构型原子间的电子自旋密度值Tab.2 Spin density between atom and atom in cluster Mn3BP

图2 团簇Mn3BP各优化构型的电子自旋密度分布图Fig.2 Spin density distribution map of each configuration in cluster Mn3BP
第二类:多重度不同几何构型很相似的构型3(4)和3(2),还有多重度不同几何构型相同的构型5(4)和2(2)。构型3(4)的B-Mn、P-Mn、Mn-Mn原子间成键时各有一个为α电子过剩,两个为β电子过剩;构型3(2)的B-Mn原子间成键时有两个为α电子过剩,一个为β电子过剩,P-Mn原子间成键时有一个为α电子过剩,两个为β电子过剩,Mn-Mn原子间成键时都为β电子过剩;以上分析说明了构型3(4)的电子自旋密度分布比构型3(2)的分布均匀。再结合自旋密度分布图分析,构型3(4)的α电子、β电子及两者重叠部分分布较构型 3(2)的更均匀,也证明了构型3(4)的电子自旋密度分布更为均匀,使构型的能量降低,更加的稳定。构型 5(4)和 2(2)构型作为几何构型相同的构型有许多相似之处,构型5(4)的B与Mn1、Mn2原子间电子自旋密度值相等(B-Mn1:-0.045;B-Mn2:-0.045),P与Mn1、Mn2原子间电子自旋密度值相等(P-Mn1:-0.056;P-Mn2:-0.056),Mn3与Mn1、Mn2原子间电子自旋密度值相等(Mn1-Mn3:0.028:;Mn2-Mn3:0.028);构型 2(2)的 B 与Mn2、Mn3原子间电子自旋密度值相等(BMn2:-0.068;B-Mn3:-0.068),P与Mn2、Mn3原子间电子自旋密度值相等(P-Mn2:-0.041;PMn3:-0.041),Mn1与Mn2、Mn3原子间电子自旋密度值相等(Mn1-Mn2:-0.007;Mn1-Mn3:-0.007);以上描述说明了构型 5(4)和 2(2)外部电子自旋密度分布比较均匀,但结合图2来看构型5(4)的原子间电子自旋密度分布要比构型2(2)均匀,且对称性更好,所以构型5(4)就能量较低,更加稳定。总结以上说法可得出结论,几何构型相同或相似但多重度不同的构型四重态的构型更加稳定。
第三类:自旋密度分布不是很明显的构型,包括构型4(4)、1(2)和 4(2)。构型 4(4)的 B与Mn2 和 Mn3原子间电子自旋密度值相等(B-Mn2:0.018;BMn3:0.018),P与Mn2、Mn3原子间电子自旋密度值相等(P-Mn2:-0.039;P-Mn3:-0.039),Mn1与Mn2、Mn3原子间电子自旋密度值相等(Mn1-Mn2:0.005;Mn1-Mn3:0.005),而其余两种构型的原子间自旋密度值没有相等的,说明构型4(4)的原子间电子自旋密度分布比较均匀,再结合图2来看构型4(4)的电子自旋密度分布的对称性比较好,所以构型 4(4)能量低,稳定性好。构型 1(2)和 4(2)相比,构型4(2)的电子自旋密度分布比构型 1(2)分布均匀,对称性也比构型 1(2)好,但是实际上构型 1(2)的能量更低,稳定性好,说明电子自旋密度只是影响构型稳定性的重要因素之一,还有其它因素也影响构型稳定性。
3 结论
团簇Mn3BP的优化构型有:戴帽三角锥(1(4)和4(4)),三角双锥(1(2)、2(2)和 5(4)),平面四边形(3(2)和3(4))和平面五边形(4(2)和2(4));各构型中(除了构型1(4)外)的非金属原子B、P上的电荷分布都是自旋向下的β电子,B-P原子间电子自旋密度均为正值,α电子过剩,B-P原子间成键时电子分布受金属Mn原子影响;几何构型相同或相似多重度不同的构型,四重态电子自旋密度分布均匀,对称性较好,能量较低,稳定性好;电子自旋密度是影响构型稳定性的因素之一,还有其它因素也影响构型稳定性。
参考文献:
[1]LANG S M,BERNHARDT T M.Gas phase metal cluster model systems for heterogeneous catalysis.[J].Physical Chemistry Chemical Physics,2012,14(26):9255-9269.
[2]KULKARNI A,LOBOLAPIDUS R J,GATES B C.Metal clusters on supports:synthesis,structure,reactivity,and catalytic properties.[J].Chemical Communications,2010,46(33):5997-6015.
[3]SHANG L,DONG S,NIENHAUS G U.Ultra-small fluorescent metal nanoclusters:synthesis and biological applications[J].Nano Today,2011,6(4):401-418.
[4]MATHEW A,PRADEEP T.Noble metal clusters:applications in energy,environment,and biology[J].Particle&Particle Systems Characterization,2015,31(10):1017-1053
[5]BONO D,BAKHAREV O N,SCHNEPF A,et al.Magnetization studies of superconductivity in a molecular metal cluster compound[J].ZeitschriftFürAnorganische Und AllgemeineChemie,2010,633(13-14):2173-2177.
[6]SOUZA C A C D,BOLFARINI C,BOTTA W J,et al.Corrosionresistanceandglassformingabilityof Fe47Co7Cr15M9Si5B15Y2(M=Mo,Nb)amorphous alloys[J].Materials Research,2013,16(6):1294-1298.
[7]CHENG Z,XU J,ZHONG H,et al.Amorphous Co-B/Al nanocomposites prepared by electroless coating technique and their excellent catalytic activity[J].Rare Metals,2010,29(1):32-36.
[8]ZHANG J,YAO H R,LI H B,et al.Amorphous alloy ni-fe-b:preparation,characterization and its catalytic ability for hydrogen generation[J].Applied Mechanics&Materials,2014,672-674:592-596.
[9]YUAN H K,CHEN H,KUANG A L,et al.Structural and magnetic properties of small 4d transition metal clusters:role of spin-orbit coupling[J].Journal of Physical Chemistry A,2012,116(47):11673-11684.
[10]GUO P,ZHENG J,GUO X,et al.Electronic and magnetic properties of transition-metal-doped sodium superatom clusters:TM@Na 8,(TM=3d,4d and 5d transition metal)[J].Computational Materials Science,2014,95:440-445.
[11]LIU D,CHEN Z,HUANG W,et al.Metal clusters constructed by dihydrazide ligand:Synthesis,structure and magnetic properties[J].InorganicaChimicaActa,2013,400(5):179-183.
[12]LIANG W,JIA J,LV J,et al.Electronic structure,stability and magnetic properties of small M 1-2 Cr(M=Fe,Co,and Ni)alloy encapsulated inside a(BN)48,cage[J].Physics Letters A,2015,379(30-31):1715-1721.
[13]BONO D,BAKHAREV O N,SCHNEPF A,et al.Magnetization studies of superconductivity in a molecular metal cluster compound[J].ZeitschriftFürAnorganische Und Allgemeine Chemie,2010,633(13-14):2173-2177.
[14]CUERVA M,GARCÍA-FANDIÑO R,VÁZQUEZVÁZQUEZ C,et al.Self-assembly of silver metal clusters of small atomicity on cyclic peptide nanotubes[J].ACS nano,2015,9(11):10834-10843.
[15]LI L,LARSEN A H,ROMERO N A,et al.Investigation of catalytic finite-size-effects of platinum metal clusters[J].Journal of Physical Chemistry Letters,2013,4(1):222-226.
[16]GIRI S,CHAKRABORTY A,CHATTARAJ P K.Potential use of some metal clusters as hydrogen storage materials—a conceptual DFT approach[J].Journal of Molecular Modeling,2011,17(4):777-784.
[17]RODUNER E,JENSEN C.Magnetic properties and the superatom character of 13-atom platinum nanoclusters[J].Magnetochemistry,2015,1(1):28-44.
[18]PAGLIONE J,GREENE R L.High-temperature superconductivity in iron-based materials[J].Nature physics,2010,6(9):645-658.
[19]SUN W,DU Y,LIU S,et al.Thermodynamic assessment of the Mn-B system[J].Journal of Phase Equilibria&Diffusion,2010,31(4):357-364.
[20]KUPCZYK A,ŚWIERCZEK J,HASIAK M,et al.Microstructure and some thermomagnetic properties of amorphous Fe-(Co)-Mn-Mo-B alloys[J].Journal of Alloys and Compounds,2018,735:253-260.
[21]ZHENG Z G,ZHU Z R,YU H Y,et al.Large magnetic entropy change and magnetic phase transitions in rapidly quenched bulk Mn-Fe-P-Si alloys[J].Journal of Alloys and Compounds,2017,725:1069-1076.
[22]REDDI P D,MUKHOPADHYAY N K,MAJUMDAR B,et al.Synthesis of Fe-Si-B-Mn-based nanocrystalline magnetic alloys with large coercivity by high energy ball milling[J].Bulletin of Materials Science,2014,37(4):815-821.
[23]NOURY S,COLONNA F,SAVIN A,et al.Analysis of the delocalization in the topological theory of chemical bond[J].Journal of Molecular Structure,1998,450(1-3):59-68.
[24]ZHANG L,YING F,WU W,et al.Topology of electron charge density for chemical bonds from valence bond theory:a probe of bonding types[J].Chemistry-A European Journal,2009,15(12):2979-2989.
[25]ORIO M,PANTAZIS D A,NEESE F.Density functional theory[J].Photosynthesis research,2009,102(2-3):443-453.
[26]HAY P J,WADT W R.Ab initio effective core potentials for molecular calculations.Potentials for K to Au including the outermost core orbitals[J].The Journal of Chemical Physics,1985,82(1):299-310.
[27]FANG Z G,HU H Z,GUO J X.Quantum chemical study on geometry and property of cluster Ni4P[J].结构化学,2006,25(1):7-16.
猜你喜欢
辽宁科技大学学报(2022年5期)2023-01-04
山西大学学报(自然科学版)(2022年5期)2022-11-23
军民两用技术与产品(2022年1期)2022-06-01
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
少儿科学周刊·儿童版(2021年22期)2021-12-11
中学生数理化(高中版.高考理化)(2021年12期)2021-03-08
原子与分子物理学报(2020年5期)2020-03-17
考试周刊(2018年39期)2018-04-19
北京航空航天大学学报(2017年10期)2017-04-20
