
中国野生小鼠来源1号染色体替换系缺失突变的发掘及功能注释
2017-03-13 06:14徐福意晁天柱胡世贤李凯周宇荀周梁良肖君华
中国实验动物学报 2017年1期
徐福意,晁天柱,胡世贤,李凯,周宇荀,周梁良,肖君华
(东华大学生物科学与技术研究所,上海 201620)
中国野生小鼠来源1号染色体替换系缺失突变的发掘及功能注释
徐福意,晁天柱,胡世贤,李凯,周宇荀,周梁良,肖君华*
(东华大学生物科学与技术研究所,上海 201620)
目的 基于野生小家鼠来源1号染色体替换系群体(population of specific chromosome 1 substitution strains, PCSSs)中18个品系的全基因组重测序结果,鉴定1号染色体上的缺失突变并对其进行功能注释。方法 采用Illumina二代测序平台获取18个品系的全基因组序列信息,通过SpeedSeq软件鉴定缺失突变,进一步利用SnpEff软件完成功能注释。结果 在18个品系的1号染色体上共检测到13 803个缺失突变。缺失长度从51 bp到70 kb不等,其中长度<500 bp的缺失突变约占总数的50%。多数缺失突变位于内含子区(50.361%)和基因间隔区(28.745%)。发现31个蛋白编码基因含有功能性缺失,其中有3个基因和人类疾病相关,7个基因参与了11条KEGG通路。结论 PCSSs的1号染色体上含有丰富的缺失突变,是在研究复杂性状的重要遗传标记。
缺失突变鉴定;二代测序;功能注释;PCSSs
现有近交系小鼠品系匮乏的遗传多样性是其在多基因复杂性状研究中进展缓慢的主要原因[1]。以具有丰富遗传多样性的野生小家鼠为供体建立的PCSSs,克服了近交系小鼠在数量性状基因座(quantitative trait locus,QTL)研究中的局限性,是QTL精确定位、基因鉴定的理想资源[2]。缺失突变作为结构变异的一种重要类型,其大小从1 bp到几十kb不等,广泛分布于人和小鼠基因组中[3,4]。研究表明,缺失突变与许多人类疾病相关[3,5]。在小鼠基因组中,缺失突变已被证明和血清尿素浓度等[6]性状相关。但迄今,PCSSs的野生小家鼠来源1号染色体中缺失突变的遗传多态性未被解析。
随着二代测序技术的不断发展,低成本、高通量的全基因组测序得以实现。通过全基因组测序,不仅可以检测单个位点的遗传突变,如单核苷酸多态性,也可以实现全基因组范围内复杂的结构变异检测,包括插入、缺失、倒位、重复等。目前,针对二代测序数据检测结构变异主要有5种策略[7]:(1)Read pair(也称为Pair-end Mapping,简称PEM);(2)Split read(简称SR);(3)Read depth(简称RD);(4)基于de novo组装的方法;(5)结合上述方法的综合策略(combinatorial approach,CA)。而SpeedSeq软件内置的lumpy-sv结合了结构变异检测的前三种策略,具有较高的灵敏度和准确度,显著优于其他软件,广泛应用于结构变异的检测[8]。
本研究以18个“野生小家鼠来源1号染色体替换系”品系为样本,采用Illumina二代测序平台获取其全基因组序列信息。利用SpeedSeq 和SnpEff 软件完成1号染色体上缺失突变的挖掘及其功能注释,全面了解和评估缺失突变在这一群体中的分布特征,进一步完善该群体的遗传变异信息,为该群体用于相关遗传学研究奠定了基础。
1 材料与方法
1.1 实验动物
实验动物来源于本实验室自建的野生小家鼠来源1号染色体替换系。动物实验遵守1988年动物管理条例,并得到东华大学实验动物委员会批准。实验在东华大学生物科学与技术研究所屏障动物实验设施进行[ SYXK (沪) 2014- 0022]。收集18个成年1号染色体替换系的鼠尾组织,-20℃保存备用。
1.2 DNA提取和测序
DNA提取采用Axygen(爱思进生物技术有限公司)基因组DNA抽提试剂盒。以0.8%琼脂糖凝胶电泳、全自动紫外与可见光分析仪FR-200A(上海复日科技实验技术研究所)和NanoDrop 2000c超微量分光光度计(Thermo Fisher Scientific, 美国)确定DNA质量和浓度。采用随机打断方式,选取大小为400~500 bp的DNA片段建库。以读长(reads)为150 bp的Illumina 高通量双末端(pair-end)测序平台(药明康德新药开发有限公司)完成全基因组重测序。
1.3 缺失突变检测及功能注释
首先,采用NGS QC软件[9]对原始数据(fastq文件)进行质控,保留质量值Q>20碱基数占整条reads碱基数>70%的reads;其次利用SpeedSeq软件[10]中的align模块将高质量的数据比对到C57BL/6J小鼠参考基因组(GRCm38/mm10),并生成bam文件;之后通过SpeedSeq软件中的sv模块鉴定缺失序列,获得初步的缺失数据集;最后,过滤掉质量值低于100、SU(support reads)<4、长度>50 bp以及鉴定为缺失一个拷贝的数据,最终生成高质量的缺失数据集。
缺失突变的功能注释利用SnpEff软件[11]完成,基因注释信息版本为Ensembl 82。此外,利用MGI中的Human-Mouse: Disease Connection 数据库[12]和DAVID: Functional Annotation Tools[13]分别完成人类疾病基因注释和KEGG 通路注释。
2 结果
2.1 测序
测序结果表明,18个1号染色体替换系均获得了大量、有效、高质量的测序数据,适用于后续生物信息学研究。如表1所示,18个样本测序数据量均超过90 Gb;质控后的高质量数据比对到参考基因组的数据百分比均在99%以上;各品系的平均测序深度均达到或高于30倍。18个样本的原始数据(fastq 文件)均已上传到NCBI (National Center for Biotechnology Information) 的SRA(Sequence Reads Archive)数据库(编号:SRP066591)。
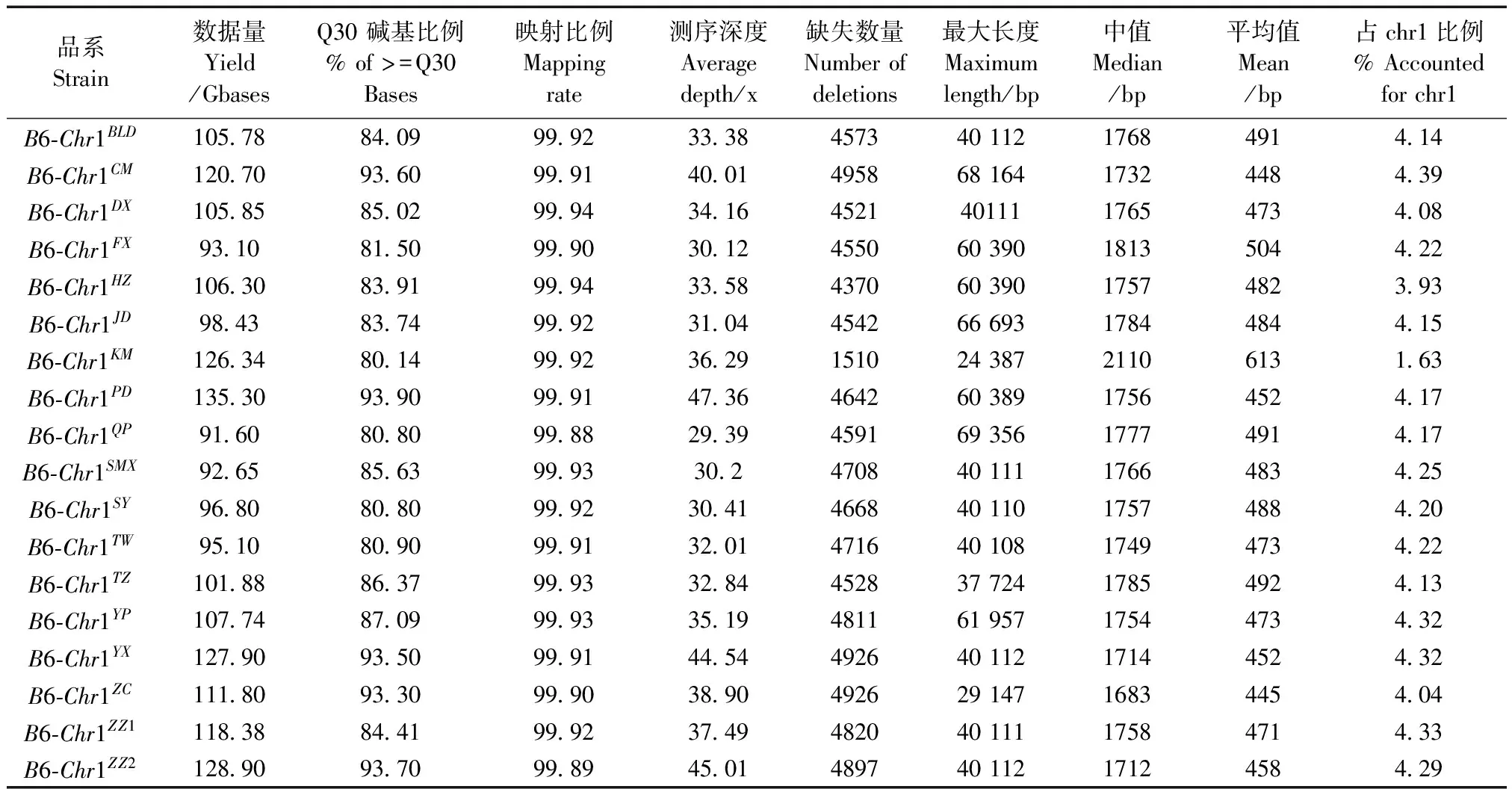
表1 18个品系DNA测序及1号染色体缺失检测统计
2.2 缺失检测

从长度分布来看,有超过半数的缺失小于500 bp(51.9%,7164个);长度在0.5~1 kb和1~2 kb的缺失分别占15.5%和11.6%;而大于10 kb的只有122个,说明缺失突变数量与缺失长度呈负相关。但长度在6~7 kb的有972个,占整体数量的7%,高于其他相近长度(图1)。此外,对缺失突变在18个小鼠品系中的频率分布统计发现(图2),约40%的缺失频率小于0.1,即这些缺失仅存在于单个品系中;另近1300个缺失频率在0.9~1.0之间,即同时存在于17或18个品系中;频率在0.5~0.6之间的最少,仅占总数的2%(278个)。

图1 缺失长度频率分布Fig.1 Size distribution of the identified deletions
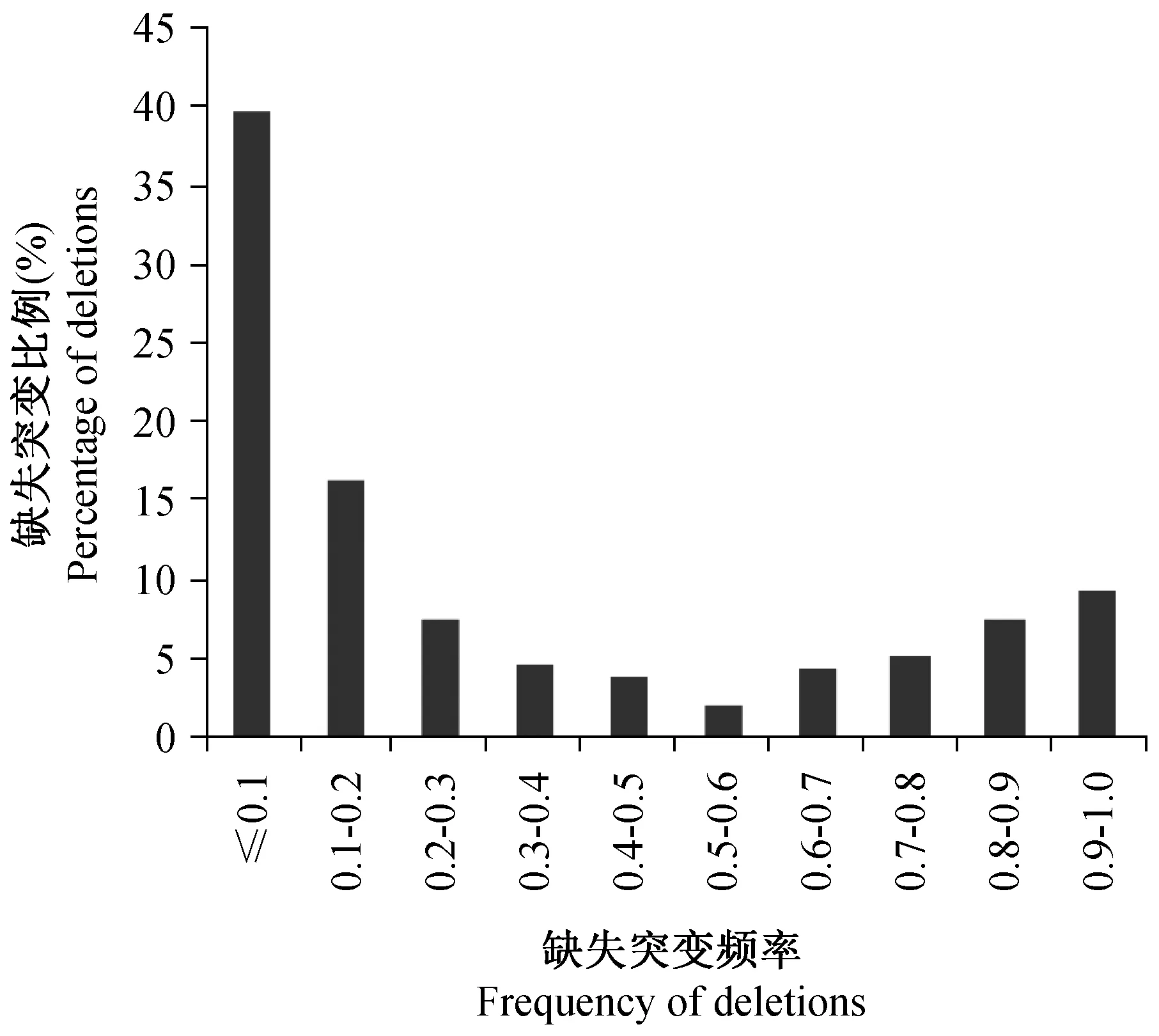
图2 缺失在18个品系中的频率分布Fig.2 Frequency distribution of identified deletions among 18 lines
2.3 功能注释
功能注释结果如表2所示,多数缺失位于内含子区(49.655%)和基因间隔区(27.955%),其次为基因上下游,其余区域所占比例均接近或小于1%。共发现605个缺失对基因功能可能产生功能性的影响(loss of function deletion),其中包括99个外显子缺失、15个移码突变、188个剪切受体或供体突变、4个起始或终止密码子突变以及199个转录本ablation。这605个缺失突变共计影响205个基因,其中包括31个蛋白编码基因(其中21个基因含有外显子缺失突变)、9个LincRNA、10个miRNA、3个snoRNA和7个snRNA。此外,还有部分缺失位于3’ UTR和5’ UTR区(0.36%),这些缺失突变可能通过影响基因表达调控序列而发挥作用。
2.4 蛋白编码基因疾病与通路注释
31个含缺失突变的蛋白编码基因的注释结果显示,其中3个基因与人类疾病相关,分别为Gigyf2,Ptpn14和Cfh。如图3中所示,在B6-Chr1QP和B6-Chr1YX小鼠中,与人类帕金森病相关(OMIM:607688)的Gigyf2基因,其第28个外显子含有一634 bp的缺失。与后鼻孔闭锁和淋巴水肿相关(OMIM:613611),属于蛋白酪氨酸磷酸酶家族的Ptpn14基因,其第3个外显子完全丢失(B6-Chr1BLD小鼠)。而与补体因子H缺乏(OMIM:126700)、溶血性尿毒综合征(OMIM:235400)以及年龄相关性
黄斑变性(OMIM:610698)有关的Cfh基因的剪切受体区域存在序列缺失。31个基因的KEGG 通路注释结果如表3所示,共发现7个基因参与了11条代谢通路,包括补体和凝血级联、药物代谢、嗅觉转导以及细胞因子-细胞因子受体相互作用等。

表2 缺失突变功能注释结果

注:图中上部分为Gigyf2基因示意图,下部分为B6-Chr1QP和B6-Chr1YX品系在Gigyf2基因上的缺失突变。图3 Gigyf2基因在B6-Chr1QP和B6-Chr1YX小鼠中的缺失突变Note. Show in upper panel is part of Gigyf2 gene model based on Ensembl,lower panel are deletions in B6-Chr1QPand B6-Chr1YXlines.Fig.3 An exon loss of Gigyf2 gene in B6-Chr1QPand B6-Chr1YXlines
3 讨论
18个1号染色体替换系小鼠的基因组中,除1号染色体来源于不同供体小鼠外,其他基因组背景均来源于近交系品系C57BL/6J。因此,本研究只针对1号染色体进行缺失的鉴定与分析。此外,因18个小鼠品系已近交化,因此,不可能出现单拷贝缺失,利用这一特征,我们对鉴定的缺失做了进一步的过滤,最终获得了高质量的缺失数据集。
作为遗传变异的一种类型,缺失可通过诸如外显子缺失、移码框改变等方式改变蛋白结构,使其丧失原有功能,也可通过改变基因拷贝数或基因转录调控序列而影响其表达量。本次研究,在18个品系的1号染色体上注释到605个功能性缺失,共影响205个基因,其中大部分基因为假基因,只有31个为蛋白编码基因。通过对31个基因做进一步的疾病与通路注释,发现3个基因与人类疾病相关,7个基因参与了11条已知通路,提示这些基因的相关表型可能在该群体中存在差异。此外,我们还发现了一些lincRNA、miRNA等存在功能性的缺失。作为非编码RNA,二者在基因转录调控方面发挥着重要作用[14,15]。

表3 KEGG 通路注释结果
中国野生小家鼠包含两个亚种M.m.musculus和M.m.castaneus[16],而KM鼠是上世纪50年代引入的Swiss小鼠,与现有近交系小鼠来源于同一亚种M.m.domesticus[17],本研究结果揭示,B6-Chr1KM品系1号染色体上缺失突变的数量(1510个)与其他17个品系相差较远,说明野生小家鼠缺失突变的多样性高于实验室小鼠。
总之,本次研究,利用二代测序技术,结合生物信息学分析手段准确解析了18个1号染色体替换系缺失突变的遗传多样性,进一步完善了该群体的遗传变异信息,为该群体用于相关遗传学研究奠定了基础。
[1] Flint J, Valdar W, Shifman S, et al. Strategies for mapping and cloning quantitative trait genes in rodents[J]. Nat Rev Genet,2005, 6(4):271-286.
[2] Xiao J, Liang Y, Li K, et al. A novel strategy for genetic dissection of complex traits: the population of specific chromosome substitution strains from laboratory and wild mice[J]. Mammalian Genome, 2010, 21(7-8):370-376.
[3] Stankiewicz P, Lupski JR.Structural variation in the human genome and its role in disease[J]. Ann Rev Med,2010, 61(1):437-455.
[4] Yalcin B, Wong K, Bhomra A, et al. The fine-scale architecture of structural variants in 17 mouse genomes[J], Genome Biol, 2012, 13(3):405-413.
[5] Weischenfeldt J, Symmons O, Spitz F, et al. Phenotypic impact of genomic structural variation: insights from and for human disease[J]. Nat Rev Genet, 2013, 14(2):125-138.
[6] Keane TM, Wong K, Adams DJ, et al. Identification of structural variation in mouse genomes.[J]. Front Genet,2014, 5:192.
[7] Zhao M, Wang Q, Wang Q, et al. Computational tools for copy number variation (CNV) detection using next-generation sequencing data: features and perspectives[J]. BMC Bioinformatics, 2013, 14(S11):1-16.
[8] Layer RM, Chiang C, Quinlan AR, et al. LUMPY: a probabilistic framework for structural variant discovery[J]. Genome Biol,2012, 15(6):1-19.
[ 9 ] Patel RK, Jain M. NGS QC Toolkit: a toolkit for quality control of next generation sequencing data[J]. PLoS One, 2012, 7(2): e30619
[10] Chiang C, Layer RM, Faust GG, et al. SpeedSeq: ultra-fast personal genome analysis and interpretation[J]. Nat Methods, 2015, 12(10):966-968.
[11] Cingolani P, Platts A, Wang LL, et al. A program for annotating and predicting the effects of single nucleotide polymorphisms, SnpEff: SNPs in the genome of Drosophila melanogaster strain w1118; iso-2; iso-3[J]. Fly, 2012, 6(2):80-92.
[12] Eppig JT, Blake JA, Bult CJ, et al. The Mouse Genome Database (MGD): facilitating mouse as a model for human biology and disease[J]. Nucleic Acids Res,2014, 43(D1):726-736.
[13] Huang DW, Sherman BT, Lempicki RA. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources[J]. Nat Protoc, 2009, 4(1):44-57.
[14] Ulitsky I, Bartel DP. lincRNAs: Genomics, evolution, and mechanisms[J]. Cell, 2013, 154(1):26-46.
[15] Jonas S, Izaurralde E. Towards a molecular understanding of microRNA-mediated gene silencing[J]. Nat Rev Genet,2015, 16(7):421-433.
[16] Boursot P, Auffray JC, Brittondavidian J, et al. The evolution of house mice[J]. Ann Rev Ecol Syst,1993, 24(4):119-152.
[17] 岳秉飞, 刘双环, 刘殿峰,等. 五个昆明小鼠封闭群遗传生化位点比较研究[J]. 实验动物科学与管理, 2003, 20(z1):58-62.
Deletion detection and functional annotation of chromosome 1 substitution strains from Chinese wild mice
XU Fu-yi, CHAO Tian-zhu, HU Shi-xian, LI Kai, ZHOU Yu-xun, ZHOU Liang-liang, XIAO Jun-hua*
(Institute of Biological Sciences and Biotechnology, Donghua University, Shanghai 201620, China)
Objective Deletion detection and annotation of 18 lines from the population of specific chromosome 1 substitution strains (PCSSs)derived from Chinese wild mice based on whole genome re-sequencing data. Methods Whole genome re-sequencing of the 18 lines were performed on the Illumina Hiseq platform. SpeedSeq software was used to detect the deletion after read alignment. Further annotation was obtained using SnpEff software. Results 13 803 deletions were identified among the 18 lines, the length of deletion was ranged from 51bp to 70 kb, among them nearly 50% were less than 500 bp. Through functional annotation,we found most of the variants were located in intronic (50.361%) and intergenic (28.745%) regions. However, we also identified 31 protein coding genes harboring loss-of-function deletions. Among them, 3 genes were associated with human diseases, 7 genes were participated in 11 KEGG pathways. Conclusion The chromosome 1 of PCSSs harbors abundant deletion mutations which can be used as genetic markers in genetic studies.
Deletion detection; Next-generation sequencing;Function annotation; PCSSs
XIAO Jun-hua,E-mail: xiaojunhua@dhu.edu.cn
国家自然科学基金面上项目(编号:31371257);上海市科技支撑计划(编号:15140900500)。
徐福意(1987-),男,博士研究生,研究方向:医学分子遗传学。E-mail: xufuyiphd@gmail.com
肖君华(1968-),男,教授,研究方向:医学分子遗传学。E-mail: xiaojunhua@dhu.edu.cn
Q95-33
A
1005-4847(2017) 01-0020-05
10.3969/j.issn.1005-4847.2017.01.004
2016-06-28
猜你喜欢
军事文摘(2022年16期)2022-08-24
今日农业(2021年11期)2021-08-13
今日农业(2020年14期)2020-12-14
中国生殖健康(2020年4期)2020-12-09
中西医结合肝病杂志(2020年2期)2020-10-27
现代农村科技(2020年9期)2020-09-08
陕西农业科学(2019年3期)2019-04-09
云南农业科技(2019年2期)2019-04-04
科学之谜(2019年3期)2019-03-28
科学之谜(2018年8期)2018-09-29
