
药物临床试验造假调查
2017-03-11 21:41邱锐
凤凰周刊 2017年1期
邱锐

过去的一年半时间,内地制药界经历了一场“大风暴”般的洗礼。
以2015年7月22日印发《关于开展药物临床试验数据自查核查工作的公告》为标志,国家食品药品监督管理总局(以下简称“食药监总局”)以“最严谨的标准、最严格的监管、最严厉的处罚、最严肃的问责”为要求,展开药品临床数据自查核查行动。被业内称为中国医药史上,监管部门对药物研发过程中临床试验数据问题的“最严核查令”。
截至2016年10月30日,食药监总局核查中心共组织检查员681人次,组成81个检查组,对分布在319家药企的148个新药注册申请品种进行了核查。
“风暴”过后的景象可以用惨烈来形容。截至2016年6月底,企业经自查主动申请撤回了1193个新药,占应自查总数的83%;2016年10月,食药监总局药品审评中心共承办新的药品注册申请中,已受理的有209个,创全年最低水平,而上一年同期数量则为548个,且这一数字还是当年的最低峰。因为监管部门大幅收紧的药物临床试验质量审查标准,大陆药企如今已经不再像从前一样,随意提交新药注册申请,而是开始变得谨慎、小心。
藥物临床试验,是药物研发链条上最重要的环节之一,指药企研发一款新药后,将药品送到医院,在人体(病人或健康志愿者)上进行的药物系统性研究。通过药物临床试验,药企能够了解新药对人体的疗效和毒副作用几何等情况。也就是说,一种药物,究竟有没有疗效、安全不安全,主要靠临床试验数据来衡量。
然而在过去很长一段时间内,因为行业发展不规范、监管能力跟不上等原因,临床数据造假或不规范现象像“潜规则”一样,在中国相关行业内大量存在,且一直未有得到社会足够的关注。
虽然此次突然对药物临床试验严格审查,让内地制药界出现了阶段性的“退潮”。但是仍需看到,药物临床试验造假是一个有着“悠久历史”的问题,其中牵涉多方利益,并非一两次核查能够改变。
正如“罗生门”一样,每一方都有委屈和不满、都希望将责任推到其他机构的身上。在这种背景下,单独依靠提高监管标准与力度,毕其功于一役,大幅提高大陆临床试验质量,进而希望提高国产药质量,效果尚待观察。
撤了83% :撤回保平安
“崩盘”、“不安”……一年多来,医药行业的朋友圈里流传着许多带有这些字眼的文章,文章都指向一件事——大量药企把自己的新药注册申请撤回了。而这都源于食药监总局开展的药物临床试验数据自查核查工作。
变化始于一年半之前。2015年7月22日,食药监总局发布《关于开展药物临床试验数据自查核查工作的公告(2015年第117号)》(以下简称“第117号公告”)。该公告指出,所有已申报并在总局待审的药品注册申请人,须对已申报生产或进口的待审药品注册申请药物临床试验情况开展自查,确保临床试验数据真实、可靠,相关证据保存完整。申请人自查发现临床试验数据存在不真实、不完整等问题的,可以在2015年8月25日前向国家食品药品监督管理总局提出撤回注册申请。
对于自查出来的问题,食药监总局对申请人采取了“恩威并施”的策略。
“恩”的做法是,在食药监总局组织核查前,药品注册申请人自查发现药物临床试验数据存在真实性问题的,应主动撤回注册申请,总局只公布其名单,不追究其责任。
“威”则体现在,食药监总局宣布,将组织专家对申请人的自查材料等进行数据分析并视情况开展飞行检查,也就是“突然袭击”式的检查。
检查中被发现临床试验数据弄虚作假的,临床试验数据不完整不真实的,不仅意味着该药品种注册或不予通过,还要“追究申请人、临床试验机构、合同研究组织的责任,并向社会公开申请人、临床试验机构、合同研究组织及其法定代表人和相关责任人员。”
根据食药监总局公开的信息,当时自查核查工作涉及1622个待审药品注册申请,其中新药948个、仿制药503个、进口药171个。
这只是一个开始。此后的一年多时间里,食药监总局又接连发布《食品药品监管总局关于进一步加强药物临床试验数据自查核查的通知》、《关于药物临床试验自查情况的公告》、《关于药物临床试验数据自查核查注册申请情况的公告(2016年第171号)》等文件,督促自查核查工作的持续进行。
上述一系列的自查核查公告,并没有立刻令众多药企掀起“撤回风潮”。业内人士告诉《凤凰周刊》记者,许多药企对于撤还是不撤曾犹豫了很久。对于申报药企来说,如果选择撤回,意味着前期投入全部“打水漂”。按照食药监总局在2015年12月发布的《食品药品监管总局关于进一步加强药物临床试验数据自查核查的通知》规定,“对主动撤回的注册申请,申请人可按新的要求重新组织开展临床试验”,如果药企不想放弃注册,就要重新组织临床试验。
业内估计,目前内地一个新药生产批件的价格(即当前国内新药研发成果交易的基准价格)可能已经上涨到超过5亿,而相应的临床批件则不低于4000万至5000万。此外,国家正在提高注册成本和要求,再申报的难度无疑要比之前大大增加。以华海药业为例,在食药监总局2015年11月26日发布的公告中,其撤回数量居首,多达8个,撤回、不批准率高达90.48%。华海药业在公告中透露,上述8个药品已累计投入的研发费用约3800万元(含部分品种的欧美注册研发费用)。
撤回显然会给药企带来不小损失,但是若等到检查人员在现场核查时发现数据存在问题时,尤其是弄虚作假,不但药品再也无法注册,相关责任人也可能会面临牢狱之灾。
这并非危言耸听,食药监总局也适时发布了造假的后果。2015年12月7日,总局披露了常州制药厂有限公司申报的阿司匹林缓释片、苏州中化药品工业有限公司申报的瑞巴派特分散片、西安恩慈制药有限公司申报的头孢地尼干混悬剂等14家企业十余个药品的造假情况,并表示“明确注册申请人、药物临床试验机构和合同研究组织的相关人员的责任,涉及医疗机构的相关责任人由卫生计生部门处理,涉嫌犯罪的移交公安机关”。
另外,截至2016年9月底,食药监总局共核查117个注册申请,对其中存在真实性问题的30个作出了不予批准的决定,约占应自查核查品种的2%;对涉嫌数据造假的27个品种11个临床试验机构和合同研究组织(CRO)予以立案调查。
在这样的背景下,大部分药企最终做出了“撤回”决定。一位医药界人士告诉《凤凰周刊》记者:“先撤回吧,虽然自己也不确定有没有问题,但还是撤回保险一些。”
于是,一股“退潮”般的景象展现在世人面前。在符合公告要求的上市和进口注册申请的1622个新药品种中,除去免临床试验的193个申请,共计1429个需要进行自查的品种。截止到2016年6月底,主动撤回数量达1193个,占比83%。
未撤回的仍有25%查出了问题
对于上述景象,有媒体以《八成新药临床数据涉假》为题进行了报道。对此,食药监总局于2015年10月专门发文反驳。一位药化注册司负责人表示,不能简单地把企业主动撤回归结为数据造假。实际上,企业自查主动撤回有多种原因,有的是不符合临床试验质量管理规范,影响试验结果科学性和准确性;有的是数据不完整,不可溯源,不足以证明申报药品的安全有效;也有的数据不真实,不排除有故意造假行为。
该表态说明,食药监总局认为临床试验的问题应该分为两类:一类属于不真实,一类属于“不规范”。按照食药监总局副局长吴浈2015年7月接受媒体采访时的表述:要严格区分数据“不真实”和“不规范、不完整”两类性质不同的问题。数据不真实问题,只要属于主观故意的,必须严肃查处。
由中国药学会主办的全国一级学术刊物《中国临床药理学杂志》2013年进行的一项调查显示,在临床试验的各个环节中,有47.80%的临床试验机构在试验实施过程中违规,更有高达85.70%的机构在试验记录环节存在问题。
此外,从官方口径来看,“不规范与造假”问题也多次被提及。2001年,食药监总局药品认证管理中心检查处处长曹彩在接受媒体采访时承认:“大部分的新药临床研究仍处于不规范状态,在这方面,中药新药的临床试验问题尤为突出。”
2004年,该局安全监管司药品研究监督处一位处长曾透露,在他们以往的监督检查中发现,临床试验存在较多不规范操作和违规问题。
十年后,2015年7月27日,吴浈在部署临床数据自查工作的电视电话会议上仍然表示:“目前药物临床试验中问题比较严重,不规范、不完整问题非常普遍。”
2016年初,食药监总局在给各省、自治区、直辖市食品药品监督管理局下发的一则通知中指出:“近期,总局针对部分试验项目多、收费低的药物临床试验机构的数据进行现场核查,发现大部分试验项目存在数据不真实、不完整、不规范问题。”
此次核查发布的最新数据也表明,国内药品的临床数据真实性问题堪忧。截至 2016年9月底,食药监总局共核查117个注册申请。在这些注册申请中,发现存在真实性问题的有30个,占比25%以上。而且,这些数据得来的时间,是在大量企业主动撤回注册申请以后,继续坚持的注册申请本应在临床数据上更为扎实,也就是说,存在作假问题的比例应该比实际情况降低了。
面对追责:利益链条上的推诿与博弈
对这场“史上最严”的数据核查,业内各方普遍给予肯定。石药集团中央药物研究院副院长杨汉煜此前在接受媒体采访时表示,加强药物临床试验数据自查核查等政策,对净化市场环境、促进医药行业创新发展起到了十分重要的作用,“以前很多企业粗制滥造的药品也往上报,根本没有时间静下心好好做创新”。
中国科学院院士、上海市科协主席陈凯先在2015年12月举行的一次会议上表示,在这样的历史背景下,加强药品数据核查十分必要。北京大学药学院药事管理与临床药学系主任史录文则认为,药企撤回注册申请,说明其申报的药品本身就有问题。“如果企业有信心,就应当按照国家的要求和流程做,这样才能保证药品安全有效。”不能用药企的经济损失和公众的健康相比,也不能因此质疑政策的合理性。
不过,大面积撤回、不批准以及被迫终止,给利益相关方不可避免地带来损失:药企面临研发费用打水漂;合同研究组织不仅浪费了很多人力物力,还要承担费用损失,以及继续选择临床机构时的责任和压力。在这种形势下,所有利益相关方,与监管部门一起,彼此推诿、角力,甚至相互指责,在造假行为被曝光后,面对申请撤回所产生的经济损失,以及可能的刑事责任究竟应该由谁来承担,形成了一个“罗生门”。
目前,大陆新药研发链条上涉及的机构主要有五部分:药品生产厂家(又称申办者)、食药监总局、经食药监总局认证的药物临床试验机构(简称GCP机构)、伦理委员会(简称IRB)以及合同研究组织(简称CRO)。
在药物临床试验中,各方分工流程为:药品生产厂家要想将药品上市,必须到食药监总局申请注册。注册手续中的一项为进行药物临床试验。进行药物临床试验之前,药品生产厂家同样需要到食药监总局申请,经审查批准、签发同意后,才能進行试验。
一旦获批,药品生产厂家会拿着批文,选择适合的GCP机构进行试验,但是试验的方案须获得IRB批准。
此外,临床试验由于从设计、实验操作,到数据统计分析,再到申请批文都是一个专业性极强的过程,催生了一种专业的中介机构——合同研究组织。一些药品生产厂家也可能通过委托合同将整个流程外包给这样的组织,直接等待最终的注册批文。
当问题出现,较为复杂的流程与环环相扣的关系,让每一方都在试图将责任推到其他机构的身上。
作为监管方,食药监总局坚称问题主要出在申办者和药物临床试验机构两者身上。2014年,原食药监总局安全监管司药品研究监督处就曾表示,造成临床试验质量不佳主要是申办者对药物临床试验机构的了解不够、缺乏责任心;临床研究机构相对短缺,法规不完善、法规执行情况不好,监管力度不够。
2015年7月,吴浈在接受媒体采访时表示,临床试验数据不真实、不完整,有着深刻的历史和社会原因。不过,主要问题仍然出在药品注册申请人及其委托的临床试验机构身上。
吴浈称,一些药品注册申请人对临床试验过程不参与、不监督,有的授意或者默许临床试验造假。申请人委托的临床试验机构对试验行为没有履行相应的监督责任,有的甚至成为规避监管、弄虚作假的“教唆者”。
药企委屈多:申报时间太长了
药企方面也有自己的委屈。恒瑞医药董事长孙飘扬在不久前的一次行业会议上“申诉”:监管部门在核查问题上,要界定造假和不规范,更要分清责任,打板子要打对地方,不要都打在申报者身上。恒瑞医药作为国内研发的佼佼者,也是目前撤回数量较多的上市药企之一,其一个投资过亿元的1.1类新药(指未在国内外上市销售的药品)刚刚被撤回。
许多药企认为目前国内新药上市前的审批时间过长。以3.1类新药(指已在国外上市销售但未在国内销售的制剂及其原料药)的审批为例,根据专业医学研究组织丁香园Insight数据库的统计,申报临床的平均审评时间为27个月,申报生产的平均审评时间为34个月,审评过程占据整个新药研发时间的64%。而若开发的是同一款新药,美国FDA进行新药审批的平均时长仅为30个月,占新药研发时间的31%。
在中国和美国,新药均享受20年的专利保护期,当药品审核的过程太长时,药品进入市场后的盈利期就会大受影响。药品研发本就需要巨额投入,所以一些迟迟无法盈利的药企便在其他地方“开源节流”了。
此外,药企还对进行临床试验机构充满抱怨。中国对临床试验机构实行资格认证制度,即只有通过药监部门审批的临床机构(多为三甲医院),才可以参与临床试验。但问题在于,药物临床试验机构数量太少,与每年需要开展临床试验的项目数量严重不匹配,导致临床试验机构不堪重负,影响试验的质量、规范。
据重庆医科大学的论文《我国药物临床试验现状与对策研究》援引的2012年数据,我国拥有近6000家药物研发企业和379家认证的药物临床试验机构。而官方公布的数据显示,在2012年当年经批准开展药物临床研究的项目高达704个。
“特别是一些针对不常见疾病的药物临床试验,药企的选择就只有几家医院,恨不得要跪着求这些医院来做试验”。一位药企前工作人员告诉《凤凰周刊》记者,按照惯例,药企往往会派出监察员(CRA)对临床试验机构进行监督,但因为缺乏第三方的监督与制衡,同时又是主动请求医院接受试验,所以很难对医院的不合规行为进行监督。
医院喊冤:临床试验数据真实性难控
负责临床试验的医院,也常常抱怨自己的难处。中国医生临床任务过于繁重,再加上临床试验对于医生的待遇和职位晋升没有促进作用,所以医生并不愿意花费过多的精力管理临床试验,一般由下属医生或研究生完成,进而导致临床试验质量不可控。
比如,有医生没有及时记录数据,等试验结束后两年核查时,凭回忆填补表格。而这种没有基于原始数据的无意识行为,会造成恶劣的后果,直接影响数据真实性。一位某三甲医院的医生向《凤凰周刊》记者透露,很多医生不愿接新药临床试验的工作,因为有的药企给的报酬较少,而一旦后期使用出现问题,药监局又可能让医生和医院承担较大的责任。
此外,对于参加临床试验的病人,医生也非常难以控制。卓永清是中国外商投资企业协会药品研制与开发行业委员会(RDPAC) 执行总裁,曾经参加过多例药品临床试验。他告诉《凤凰周刊》记者,在一些地方的医院,有的病人没有参加医保,为了报销额度,会冒用亲戚朋友的身份看病。由此导致的问题是,医生根本无法掌握前来看病的患者的真实情况,进而影响临床试验的质量。
作为连接医院和药企的中介机构——合同研究组织(CRO),则成为众矢之的。由于这些机构的主要工作是帮药企完成临床试验以及繁杂的申报手续,并以此赚取服务费,因此,很多药企和临床试验机构都认为,为了更快更多地获取利益,CRO是药品临床试验造假的主谋。
CRO参差不齐的服务水平,会造成“劣币驱逐良币”的市场隐患。目前,国内CRO行业缺乏准入门槛,相关规定只要求“依照合同提供技术服务的能力”并且是“正规的法人组织”就可以,导致CRO企业数量多、规模小,恶性竞争严重。
据制药行业专业媒体《E药经理人》报道,在2016年春节后的某交流会中,食药监总局某官员直接放话:凡临床试验的合同金额低于10万元肯定造假。很多行业人士认为,这种说法在某种程度上是合理的,“几万块钱,连试验要用到的试剂都买不起,唯有系统地编造数据才能完成任务。”
但CRO机构同样认为自己很冤枉。多位曾经就职于这类机构的人士告诉《凤凰周刊》,CRO经常处在一个非常尴尬的地位:有药企认为自己已经购买了服务,所以要求CRO无论采取何种手段,必须保证临床试验结果符合标准;与此同时,一些临床试验机构又非常抢手,CRO经常需要放低身段来请求医院接受临床试验,所以对于医院的一些不合规行为,CRO也只能默许。
“严进宽出”的监管模式亟待改善
2009年,食药监总局南方经济研究所、《中国处方药》和中国外商投资协会药品研制与开发行业协会(RDPAC)联合发布的《中国药物临床试验现状和发展》调研报告明确指出,中国药物临床试验发展的最大障碍是“严进宽出”政策,即药物临床试验审批期过长、流程繁琐,但对临床试验实施和上市后药物监管不足。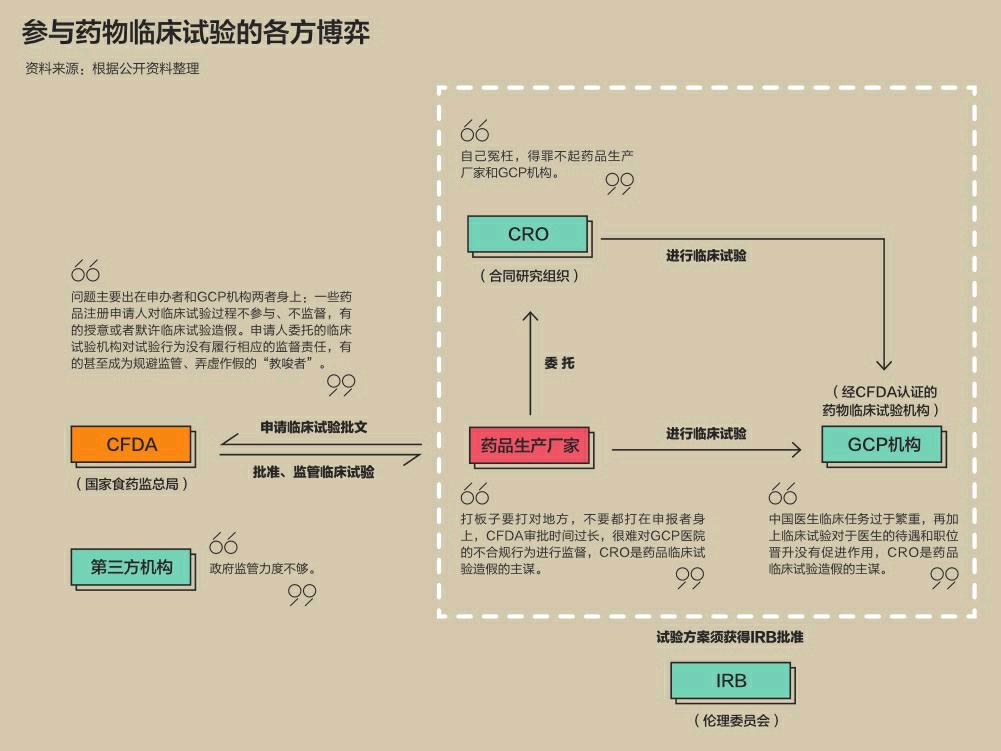
虽然2015年7月22日之后,食药监总局对存在真实性问题的30个品种作出了不予批准的决定,对涉嫌数据造假的27个品种11个临床试验机构和合同研究组织(CRO)予以立案调查,但在此之前,自 2001 年发布的《中华人民共和国药品管理法》规定,申请药品被“枪毙”、相关机构被处理之后,幾乎查不到因临床试验阶段数据造假而被公开处理的药企、医院机构或 CRO 公司名单和信息。
相互指责的“罗生门”说明了中国临床试验问题背后的原因错综复杂,也和药物临床试验现状、企业的利益博弈,以及中国药品研发的整体发展水平密切相关。由此,也让业内人士普遍认为,此次掀起的“核查风暴”,能否形成长期效果有待观察。毕竟,“运动式”的监管难以持续和常态化,若不制度化,不能触及根本的矛盾点,难以扭转现实。
2015年8月,国务院出台《关于改革药品医疗器械审评审批制度的意见》,其中提出要提高审评审批质量,严肃查处注册申请弄虚作假行为等目标和要求。这一《意见》被业内视为未来可能改变中国药物临床试验数据混乱现状的重要文件。
百瑞鼎辉医药研究有限公司总经理娄实认为,临床数据核查如何持续成为常态化监管还需要一个过程,但是中国药品研发监管的未来方向已经非常清楚,即按照全球药品临床试验管理规范(GCP)标准,围绕质量、安全性和有效性三个要素来检验中国药品的研发质量已是大势所趋。
猜你喜欢
中国药学药品知识仓库(2022年7期)2022-05-10
中国质量与标准导报(2020年5期)2020-12-08
科技风(2018年22期)2018-10-21
食品界(2018年4期)2018-06-14
现代交际(2017年7期)2017-05-31
健康管理(2016年7期)2016-05-14
决策探索(2016年4期)2016-03-28
