
Adenyl cyclase activator forskolin protects against Huntington’s disease-like neurodegenerative disorders
2017-03-09 06:05:51SidharthMehanShabaParveenSanjeevKalra
中国神经再生研究(英文版) 2017年2期
Sidharth Mehan, Shaba Parveen, Sanjeev Kalra
Department of Pharamcology, Rajendra Institute of Technology & Sciences, Sirsa, Haryana, India
Adenyl cyclase activator forskolin protects against Huntington’s disease-like neurodegenerative disorders
Sidharth Mehan*, Shaba Parveen, Sanjeev Kalra
Department of Pharamcology, Rajendra Institute of Technology & Sciences, Sirsa, Haryana, India
Long term suppression of succinate dehydrogenase by selective inhibitor 3-nitropropionic acid has been used in rodents to model Huntington’s disease where mitochondrial dysfunction and oxidative damages are primary pathological hallmarks for neuronal damage. Improvements in learning and memory abilities, recovery of energy levels, and reduction of excitotoxicity damage can be achieved through activation of Adenyl cyclase enzyme by a specif i c phytochemical forskolin. In this study, intraperitoneal administration of 10 mg/kg 3-nitropropionic acid for 15 days in rats notably reduced body weight, worsened motor cocordination (grip strength, beam crossing task, locomotor activity), resulted in learning and memory def i cits, greatly increased acetylcholinesterase, lactate dehydrogenase, nitrite, and malondialdehyde levels, obviously decreased adenosine triphosphate, succinate dehydrogenase, superoxide dismutase, catalase, and reduced glutathione levels in the striatum, cortex and hippocampus. Intragastric administration of forskolin at 10, 20, 30 mg/kg dose-dependently reversed these behavioral, biochemical and pathological changes caused by 3-nitropropionic acid.These results suggest that forskolin exhibits neuroprotective ef f ects on 3-nitropropionic acid-induced Huntington’s disease-like neurodegeneration.
nerve regeneration; Huntington’s disease; mitochondria; adenyl cyclase; forskolin; oxidative stress; basal ganglia; neural regeneration
Introduction
Huntington’s disease (HD) is a neurodegenerative disorder characterized by increasing mental disturbances, cognitive impairment, jerky movements and weight loss and involves the basal ganglia, cerebral cortex and hippocampus (Walker, 2007; Sasone et al., 2009). In HD, mitochondrial dysfunction (Costa and Scorrano, 2012; Chaturvedi and Beal, 2013), energy depletion (Ribeiro et al., 2012), oxidative stress and glutamate excitotoxicity (Kim et al., 2011), and transcriptional dysregulation (Basso, 2012) are often accompanied by neurochemical dysregulations like γ-amino butyric acid (GABA), glutamate, dopamine (DA) and adenosine receptors in basal ganglia, responsible for motor functions (Chen et al., 2013).
Mitochondrial 3-nitroproprionic acid (3-NP) can reverse behavioral, biochemical, and striatal pathological alterations found in HD-like neurotoxicity (Dhir et al., 2008; Kumar and Kumar, 2009; Liot et al., 2009; Delorme et al., 2012). 3-NP is a naturally occurring mycotoxin that is a suicidal irreversible inhibitor of succinate dehydrogenase (SDH), an enzyme present in inner mitochondrial membrane responsible for the reduction of ATP synthesis and the initiation of oxidative stress (Binawade and Jagtap, 2013; Jadiswami et al., 2014). There is strong evidence that cAMP dependent CREB phosphorylation induces long term memory (LTP), inhibits apoptotic and necrotic cell death, and suppresses the synthesis of proteins which are important for the growth and development of synaptic connection and strength (Zhang et al., 2008; Benito and Barco, 2010; Bitner, 2012). The agents that greatly enhance the cAMP/PKA/CREB signaling pathways can prevent against cerebral stroke and various neurological complications. The levels of adenyl cyclase (AC) and cAMP have been conf i rmed to be reduced under neuropathological conditions (Puzzo et al., 2005). The benef i cial ef f ects of natural AC activator Coleus Forskohlii (FSK) against various neurodegenerative abnormalities through the modulation of CREB and brain-derived neurotrophic factor (BDNF) (Heo et al., 2013; Rosales-Corral et al., 2015). cAMP-dependent pathways involve various neurotransmitters including serotonin, acetylcholine, glutamate, and dopamine (Gloerich and Bos, 2010) and plays an important role in cognitive functioning. The activation of the cAMP-dependent protein kinase (PKA) significantly inhibits the release of neuroinfl ammatory cytokines like interleukins (ILs), tumor necrosis factor-α (TNF-α) (Chong et al., 2003), and inducible nitric oxide synthase (iNOS) in astrocytes and macrophages (Pahan et al., 1997) which are implicated in neuroinf l ammation and oxidative stress (Boulanger and Poo, 1999). cAMP signaling pathway is involved in release of BDNF (Ji et al., 2005), plays an important role in neuronal survival and synaptic plasticity (Pizzorusso et al., 2000) as well as improvements in learning and memory impairment (Zhang et al., 2015). cAMP elevation reverses energy def i cits, reduces excitotoxic damage, prevents neurotoxicity (Zou and Crews, 2006),promotes biosynthesis, increases neurotransmitter release (Leenders and Sheng, 2005), inhibits apoptotic and necrotic cell death (Nishihara et al., 2003), and improves neuronal functioning (Gong et al., 2004). Preventive treatments of HD are still limited to complementary care and management of various neurological complications. The present study was designed to investigate the pathogenesis of HD and the role of forskolin (FSK) in 3-NP-induced HD-like neurodegenerative disorders.
Materials and Methods
Animals
Male Wistar rats, weighing 220–250 g, aged 8–9 months, were provided by Laboratory Animal Center, Rajendra Institute of Medical Sciences, China where animals were placed in polyacrylic cages under standard husbandry conditions at temperature 22 ± 2°C with proper food and water ad libitum.The experimental design was conf i rmed by Institutional Animal Ethics Committee (IAEC) (RITS/IAEC/2014/03/03) as per the instructions of CPCSEA, Government of India (888/ PO/Re/S/05/CPCSEA). Animals were accommodated to laboratory conditions prior to experimentation.
Drugs and treatment
3-NP was obtained from Sigma-Aldrich, St. Louis, MO, USA. FSK was from Bangladesh Petroleum Exploration and Production Company Ltd, Rajasthan, India. All chemicals used in the experiments were of analytical grade. Solutions of the drugs and chemicals were freshly prepared before use. 3-NP was dissolved in 5% dimethyl sulfoxide in saline (pH 7.4) and administered intraperitoneally (i.p.) at 10 mg/kg for 15 days. FSK was dissolved in 2% ethanol until it was soluble in water and then intragastrically (i.g.) administered. Rats were divided into six groups (n = 6 per group): normal, FSK only, 3-NP only, FSK10 + 3-NP, FSK20 + 3-NP, FSK30 + 3-NP groups. In the normal, FSK or 3-NP group, rats were treated with normal saline, FSK (30 mg/kg/d, i.g.) only or 3-NP (10 mg/kg/d, i.p.) only for 15 days. Rats in the latter three groups were subjected to FSK (10, 20, 30 mg/kg/d, i.g.) administration prior to 3-NP (10 mg/kg/d, i.p.) for 15 days (Figure 1).
Measurement of rat body weights
Rat body weight was measured on days 1 and 15 of the study. Rat body weight change was evaluated by (body weight on day 1— body weight on day 15)/body weight on day 1 × 100%.
Behavioral changes
Spatial navigation task in Morris water maze
Rat spatial learning and memory was estimated in a Morris water maze on days 5, 10 and 15 of the study. The Morris water maze is a round water pool (180 cm diameter, 60 cm height) filled with water (25 ± 1°C) to a depth of 40 cm (Morris, 1984). A non-toxic water emulsion was used to turn water from clear to turbid. Four start locations (north, south, east, west) were designated on the wall to distribute the pool into four quadrants. An escape platform (10 cm in diameter) was submerged approximately 2 cm below the water surface and placed in the center of one of four quadrants throughour the entire behavior test. Before the start of normal training, the rats were allowed to swim freely for 120 seconds without platform. Animals daily received one session of training (four trials each session; one session per day) for 4 days (days 1, 2, 3 and 4) before fi nal trail, i.e. on days 5 and 10. Both the cut-of f time and the fi nish time for each trial were 120 seconds. after reaching the hidden platform, the animals were allowed to stay there for 30 seconds before start of the next trial. If the rats were unable to fi nd out the hidden platform within 120 seconds, it was gently placed on the hidden platform and permitted to stay there for the same time period. The time taken up by the rats to fi nd the designated platform, i.e., time spent in target quadrant zone (TSTQ), was also estimated. after the acquisition phase (24 hours), a probe test (day 15) was performed by removing the hidden platform from the Morris water maze. Rats were allowed to swim freely in the maze for 120 seconds and TSTQ was recorded (Ishrat et al., 2006; Deshmukh et al., 2009).
Spontaneous locomotor activity
On days 1, 5, 10 and 15, the spontaneous locomotor activity of each rat was measured for duration of 5 minutes in a closed area equipped with infrared sensitive photocells with a modern electrical photoactometer (Company INCO, Ambala, Haryana, India). Independent interference with beam crossing induced an electric signal, which can be seen on a digital reader. The photoactometer was fi xed in a silent well ventilated place in the laboratory. Rats were evaluated over a period of 5 minutes and data were measured as counts per 5 minutes (Sharma and Gupta, 2003; Kumar et al., 2006).
String test for grip strength
The rats were granted to stay with the forepaws on a steel wire (diameter 2 mm and length 35 cm) 50 cm high over a soTh support on days 1, 5, 10 and 15. The time taken up by the rats to hold the wire was recorded. The latency to hold the grip loss is conf i rmed as a measurement of muscle grip strength (Shear et al., 1998).
Elevated plus maze (EPM) test
The EPM test (Kumar and Kumar, 2009) is a behavioral assy used to measure an animal performance in external environment, i.e. an animal’s working memeory. The EPM has two open arms and two closed arms. These arms are elevated from the middle platform and the EPM was kept up to a height of 50 cm from the fl oor. The EPM test was performed on days 13 and 14 of the designed schedule. Each rat was placed at the end of the open arm, facing against the middle platform on day 13. Transfer latency, which is the time taken up by the rat to enter one of the closed arms with its four paws, was condirmed on day 13, i.e. acquisition trail. If a rat did not enter one of the closed arms within 120 seconds, then it was gently forced into one of the two closed arms and transfer latency was noted as 120 seconds. The rat was allowed to stay in the maze for 10 seconds and then back to its main cage. The transfer latency,i.e. retention latency, was evaluated again 24 hours after the fi rst trial on day 14.
Beam crossing task
In this behavioral paradigm, rats were allowed to move across a narrow wooden beam to evaluate their motor coordination. The instrument used for beam crossining task consisted of two platforms connected by a wooden beam. The beam was elevated 50 cm above the ground. To provide protection for a falling rat, a soTh cushion was placed below the beam. At the start of the experiment on day 15, rats were allowed to stay on the beam for 5 minutes. The training trial was started by putting the rats on the platform at one end. When rats moved across the beam from one end to the other end, slipping of its paw occurred. In every trial, the number of slips by individual rats was recorded. Rat motor coordination was rated on a scale from 0 to 4. A score of 0 indicated that rats could immediately move across the beam. Scores 1, 2 and 3 were given to the rats that showed mild, moderate and severe motor dysfunctions accordingly. A score of 4 was marked to the rats that could be move on the desired platform (Singh et al., 2015).
Assessment of biochemical parameters
Brain homogenate formation
On day 15, rats were sacrificed by decapitation, and the brains were dissected and washed with ice-cold isotonic saline solution. Brain samples from the cortex, striatum and hippocampus were homogenized with 10 times (w/v) ice cold 0.1 M phosphate buffer (pH 7.4). The mixed clear solutions were then centrifuged at 10,000 × g for 15 minutes. Supernatant was taken out and aliquots were used for biochemical estimation.
Adenosine triphosphate (ATP) assessment
A portion of homogenate was sonicated sufficiently in ice cold perchloric acid (1N) to suppress the ATPase. After centrifugation (14.000 × g, 4°C, 5 minutes), supernatant containing ATP was neutralized with 1 N NaOH and placed at –80°C until evaluation ends. ATP level in supernatant was measured using reversed-phase high-performance liquid chromatography (RP-HPLC) (PerkinElmer Inc., Hopkinton, MA, USA). RP-HPLC quantification was evaluated on a reversed-phase Hypersil C18 (4.6 mm × 250 mm, 5 μm column (Elite, Dalian, China) joined to two LC-10ATvp pumps (Shimadzu, Kyoto, Japan), associated with UV-Vis detector.The mobile phase was 100 mM KH2PO4buf f er solution (pH 6.0), the fl ow rate 1.2 mL/min, the column temperature 25°C and the detection wavelength 254 nm. A reference solution of ATP was made according to dissolving standard (Sigma, St. Louis, MO, USA) (Ramanathan et al., 2012).
Assessment of brain tissue protein
The protein content was measured by biuret method using bovine serum albumin as standard (Gornall et al., 1949).
Succinate dehydrogenase (SDH) activity
SDH is a biochemical indicator of mitochondrial dysfunction in neurodegenerative disorders. The ef f ective evaluation of SDH in the brain was conf i rmed according to the procedure detailed in various results (Kumar et al., 2007). Sodium succinate solution 0.3 mL was stirred with 50 μL of remaining homogenate. The admixture was then incubated at 37°C for 10—20 minutes. Following addition of 0.1 mL of p-iodonitrotetrazolium violet (INT), the admixture was again incubated for 10 minutes. In this procedure, reaction mixtuire was inhibited by 1 mL of the mixture of ethyl acetate, ethanol and tricholoroacetic acid (5:5:1, v/v/w) and centrifuged at 15,000 rpm for 1 minute. The absorbance at 490 nm was measured with a spectrophotometer (Shimadzu, UV-1700).
Lactate dehydrogenase (LDH) activity
LDH activity in rat brain homogenate was measured using a LDH kit (Transasia Bio-Medicals Ltd., Mumbai, India) and it was expressed as IU/L (Maharaj et al., 2003; Choi and Lee, 2004).
Acetylcholinesterase (AChE) activity
AChE activity in rat brain homogenate was measured as per the procedure given by Ellman et al. (1961). The reaction solutions contained 0.05 mL of remaining residue (pH 8), 0.3 mL of 0.01M sodium phosphate buffer, 0.10 mL of acetylthiocholine iodide and 0.10 mL of 5,5’- dithiobis(2-nitrobenzoic acid (Ellman reagent). The absorbance at 412 nm was read using a UV-VIS spectrophotometer (Labindia, Maharashtra, India). AChE enzymatic activity was denoted as μmolmg protein (Ellman et al., 1961).
Reduced glutathione (GSH) levels
As per the procedure mentioned by Ellman (1959), the level of reduced glutahione in rat brain homogenate was quantified. A 1 mL of supernatant was precipitated with 1 mL of 4% sulfosalicylic acid and cold digested at 4°C for 1hour. after centrifugation at 1,200 × g for 15 minutes, 0.1 mL of supernatant, 2.7 mL of 0.1 M phosphate buf f er solution (pH 8) and 0.2 mL of DTNB were added to the mixture. Due to reaction, yellow color appeared fi rst, absorbance at 412 nm was measured using a UV-VIS spectrophotometer (Labindia) and GSH level was expressed in μmol/mg protein.
Malondialdehyde (MDA) levels
The level of oxidative stress marker MDA in rat brain homogenate was measured as per the procedure reported by Wills (1996). Absorbance at 532 nm was read using a UVVIS spectrophotometer (Labindia) and MDA level was indicated as nmol/mg protein.
Catalase activity
The activity of catalase, an anti-oxidative enzyme was detected according to the method by Aebi et al. (1974). 0.1 mL supernatant was mixed in a cuvette in which 1.9 mL of 50 mM phosphate buffer soluation (pH 7.0) was added. Reaction was initiated by adding 1.0 mL of freshly prepared 30 mM H2O2. Absorbance at 240 nm was measured using a UV-VIS spectrophotometer (Labindia). Catalase activity wasexpressed as % control.
Super oxide dismutase (SOD) activity
Antioxidant enzyme SOD activity was measured as per the method given by Misra and Fridovich (1972). Epinephrine underwent auto-oxidation at pH 10.2. 0.2 mL supernantant of rat brain homogenate, 0.8 mL of 50 mM glycine buf f er (pH 10.4) and 0.2 mL epinephrine were mixed together. Five minutes later, absorbance at 480 nm was measured using a UV-VIS spectrophotometer (Labindia). SOD activity was expressed as % control.
Nitrite levels
Nitrite level, an indicator for the release of nitric oxide, was measured by Greiss reagent (0.1% N-(1-naphthyl) ethylene diamine dihydrochloride and 1% sulfanilamide and 2.5% phosphoric acid) according to the report by Green et al. (1982). Residue mixture of rat brain homogenate and Greiss reagent were suf fi ciently mixed and then incubated at room temperature for 10 minutes. Absorbance at 540 nm was measured using a UV-VIS spectrophotometer (Labindia). Nitrite level was expressed as μmol/mg protein.
Histophathological studies
Brain samples (cortex, striatum and hippocampus) were treated with 10% formalin solution, embeeded with paraffin wax, sectioned and then stained by hematoxylin and eosin (Kumar and Kumar, 2009). After fixation with DPX 292, brain sections were observed under a light microscope (Nikon, Japan) and photographed using a highly digital zoomed camera.
Statistical analysis
All data were statistically analyzed using GraphPad Prism v5.01 (Graph pad soThware, INC, La Jolla, CA, USA) and expressed as the mean and standard deviation (SD). Observed data were analyzed using one-way analysis of variance followed by Bonferroni post hoc test or Tukey’s multiple comparisons test. A level of P < 0.05 was considered statistically signif i cant.
Results
Ef f ect of FSK on body weight of 3-NP treated rats
3-NP administration resulted in a signif i cantly decreased body weight compared with normal saline administration. Treatment with FSK only did not result in any remarkable alteration in body weight when compared with normal saline administration. Prior treatment with FSK 10, 20 and 30 mg/kg in 3-NP-treated rats signif i cantly reversed the 3-NP-mediated reduction in body weight (P < 0.05). FSK 30 mg/kg adminsitration more greatly reversed 3-NP injection-caused rat body weitght loss than FSK 10 and 20 mg/kg administration (P < 0.05; Figure 2).
Ef f ect of FSK on learning and memory abilities of 3-NP treated rats
Spatial navigation task using Morris water maze
On days 5 and 10, the transfer latency for each rat was evaluated. Obvious alaterations in transfer latency were observed in 3-NP-treated rats than in normal saline-treated or FSK only-treated rats. Transfer latency was significantly decreased in the FSK10 + 3-NP, FSK20 + 3-NP and FSK30 + 3-NP groups than in the 3-NP only group (P < 0.05). Transfer latency was signif i cantly decreased, and memory ability was signif i cantly improved in FSK 30 + 3-NP group than in FSK 20 + 3-NP or FSK 10 + 3-NP groups (P < 0.05) (Figure 3A).
On day 15 of the study, TSTQ was signif i cantly reduced in 3-NP only group than in normal group or FSK-only group (P < 0.05). There was no obvious change in TSTQ between FSK only and normal groups. TSTQ was significantly increased in FSK 10 + 3-NP and FSK 20 + 3-NP groups than in 3-NP only group (P < 0.05). Rat memory was signif i cantly improved in FSK 30 + 3-NP group than in FSK 20 + 3-NP or FSK 10 + 3-NP group (P < 0.05; Figure 3B).
EPM
On day 14, EPM transfer latency was recorded to assess rat learning and memory abilities. Transfer latency was signif icantly increased in 3-NP only group than in normal or FSK only group (P < 0.05). There was no significant difference in transfer latency between FSK only and normal groups. Transfer latency signficaintly increased in FSK30 + 3-NP, FSK 20 + 3-NP and FSK 10 + 3-NP groups than in 3-NP only group (P < 0.05; Figure 3C).
Ef f ect of FSK on motor function of 3-NP treated rats
Locomotor activity
On days 1, 5, 10 and 15, rat locomotor activity was evaluated. Rat locomotor activity was slightlty changed in 3-NP only group than in normal or FSK only group (P <0.05). There was no signif i cant dif f erence in rat locomotor activity between FSK only and normal groups (P < 0.05). Rat locomotor activity was signif i cantly dose-dependently changed in FSK30 + 3-NP, FSK20 + 3-NP, and FSK10 + 3-NP groups (P < 0.05). These results suggest that FSK exhibits remarkable ef f ects on rat locomotor activity (Figure 4A).
Grip strength
On day 1, there was no obvious dif f erence in time that rats took up to hold the wire in grip strength task between all treatment groups. On days 5, 10 and 15, the time was signif icantly reduced in the 3-NP only group than in the normal group (P < 0.05; Figure 4B). However, chronic treatment with FSK 10, 20 and 30 mg/kg greatly ameliorated 3-NP-induced reduction in grip strength (P < 0.05).
Balance motor function
On day 15, balance motor function was evaluated. The number of slips (Figure 4C) was greatly increased, i.e., the impairment in beam walking performance was signif i cantly worsened in the 3-NP only group than in the normal or FSK only groups, as conf i rmed by neurological scores (P < 0.05; Figure 4C, D).
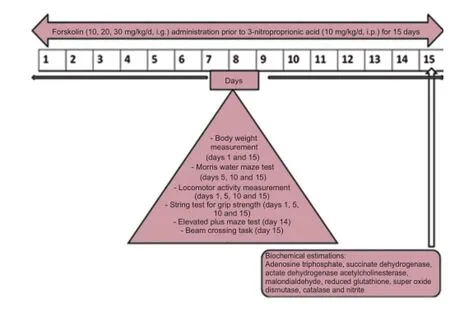
Figure 1 Drug treatment and evaluation indices.
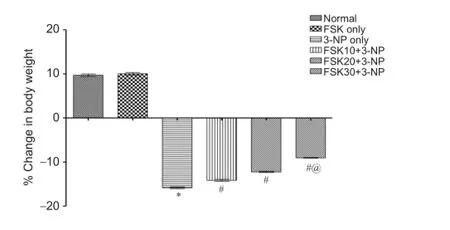
Figure 2 Ef f ect of forskolin (FSK) on body weight of 3-nitroproprionic acid (3-NP) treated rats.
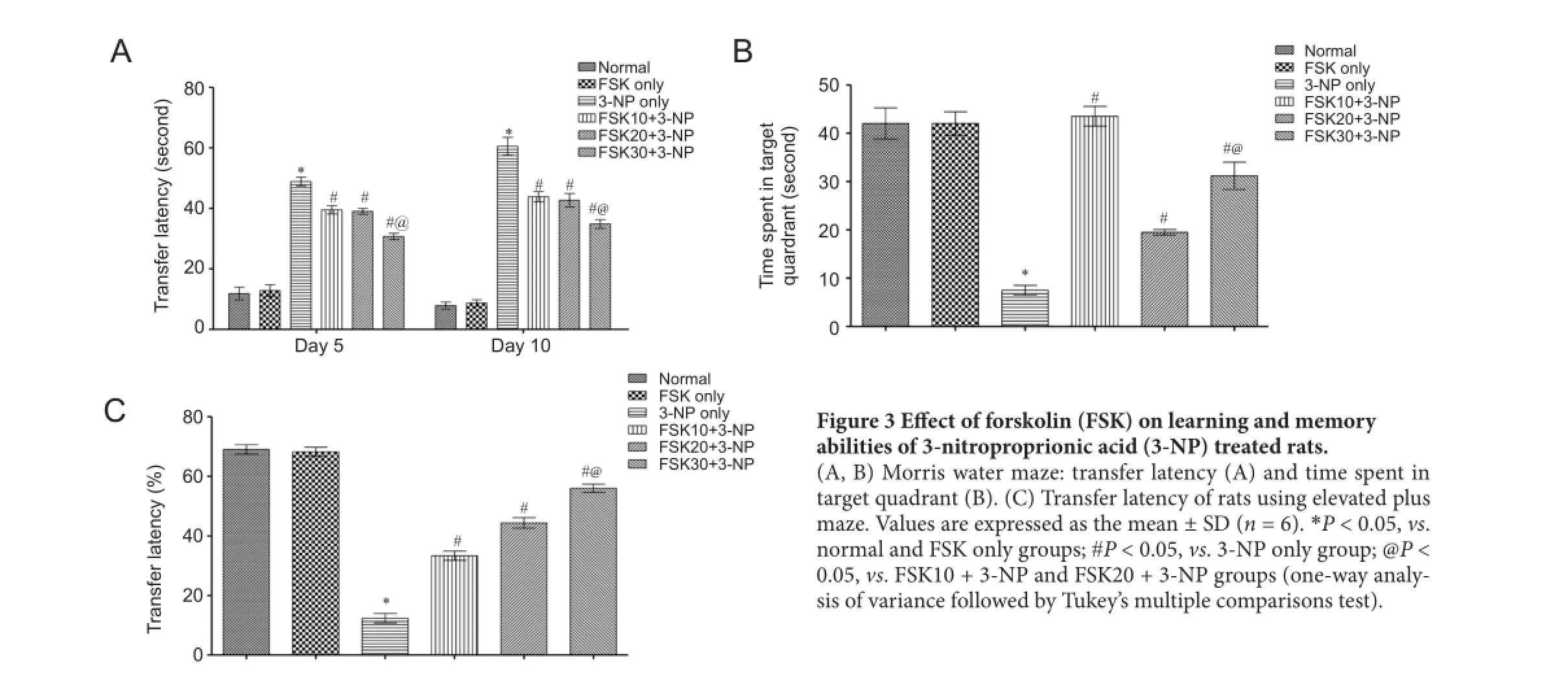
Figure 3 Ef f ect of forskolin (FSK) on learning and memory abilities of 3-nitroproprionic acid (3-NP) treated rats.
E ff ects of FSK on biochemical indices in the striatum, cortex and hippocampus of rats
ATP levels
ATP level in the homogenate of rat striatum, cortex and hippocampus was signi fi cantly decreased in the normal or FSK only group than in the 3-NP only group (P < 0.05). There was no significant difference in ATP level between normal and FSK only groups. ATP level was signi fi cantly increased in FSK10 + 3-NP, FSK20 + 3-NP and FSK30 + 3-NP groups than in the 3-NP-only groups (P < 0.05). ATP level was signfi caintly increased in the FSK 30 + 3-NP group than in the FSK 20 + 3-NP and FSK 10+3-NP groups (P < 0.05; Table 1).
SDH activity
Mitochondrial complex II SDH activity in the homogenate of rat striatum, cortex and hippocampus was significantly decreased in 3-NP only group than in normal or FSK only group (P < 0.05; Table 1). SDH activity was signi fi cantly decreased in FSK10 + 3-NP, FSK20 + 3-NP and FSK30 + 3-NP groups than in the 3-NP-only groups (P < 0.05). SDH activiaty was signif i cantly decreased in the FSK30 + 3-NP group than in the FSK20 + 3-NP and FSK10 + 3-NP groups (P <0.05).
LDH activity
LDH activity in the homogenate of rat striatum, cortex and hippocampus was significantly increased in the 3-NP only group than in the normal or FSK only group (P < 0.05; Table 1). This suggests that FSK exhibits neuroprotective ef f ects in 3-NP-treated rats. Long-term administration of FSK at 10, 20 and 30 mg/kg signif i cantly decreased LDH activiaty compared to the level in the 3-NP only group (P < 0.05). LDH activity was signif i cantly greater in the FSK30 + 3-NP group than in the FSK20 + 3-NP or FSK10 + 3-NP group (P < 0.05).
AChE activity
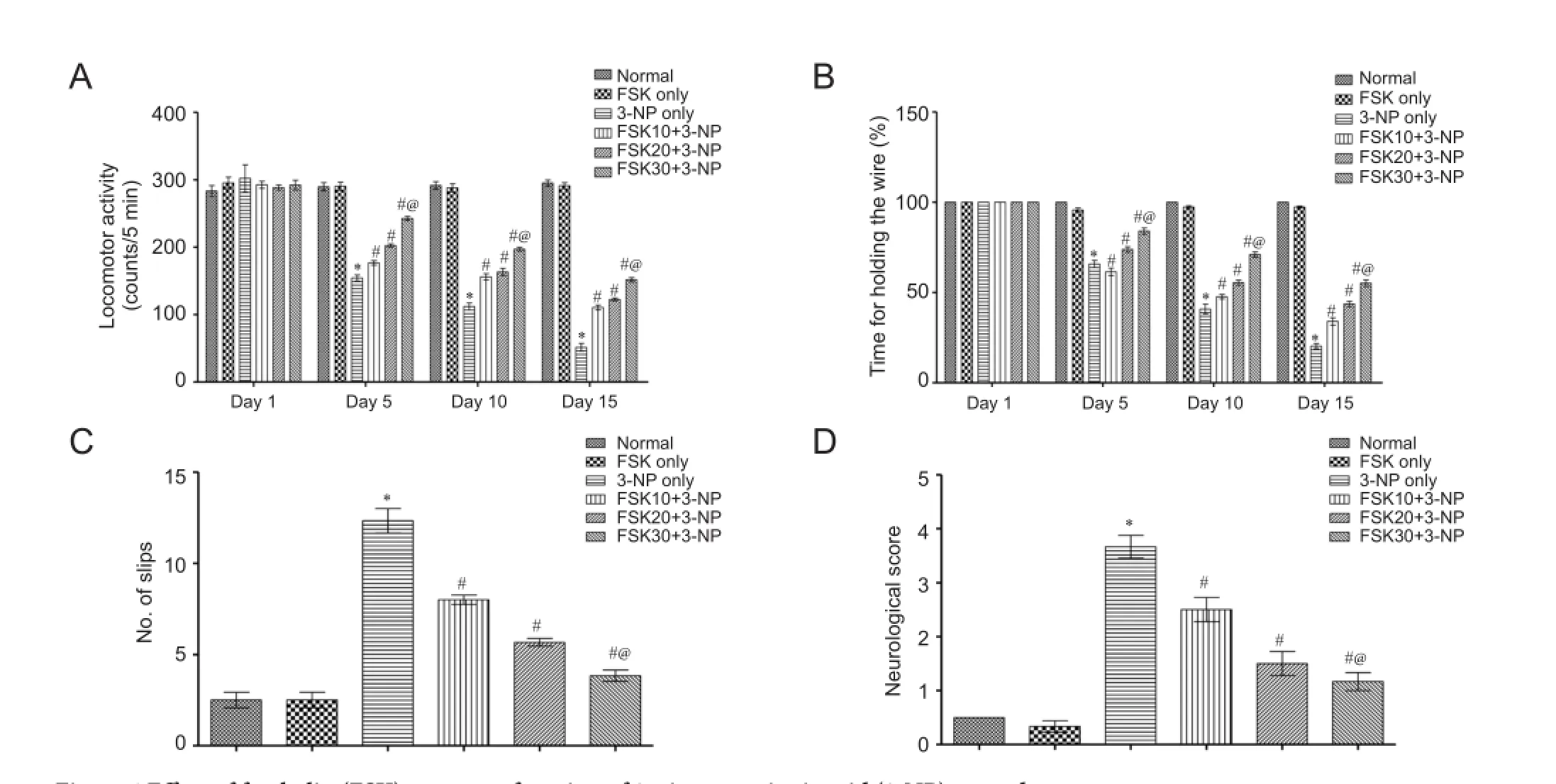
Figure 4 Ef f ect of forskolin (FSK) on motor function of 3-nitroproprionic acid (3-NP) treated rats.
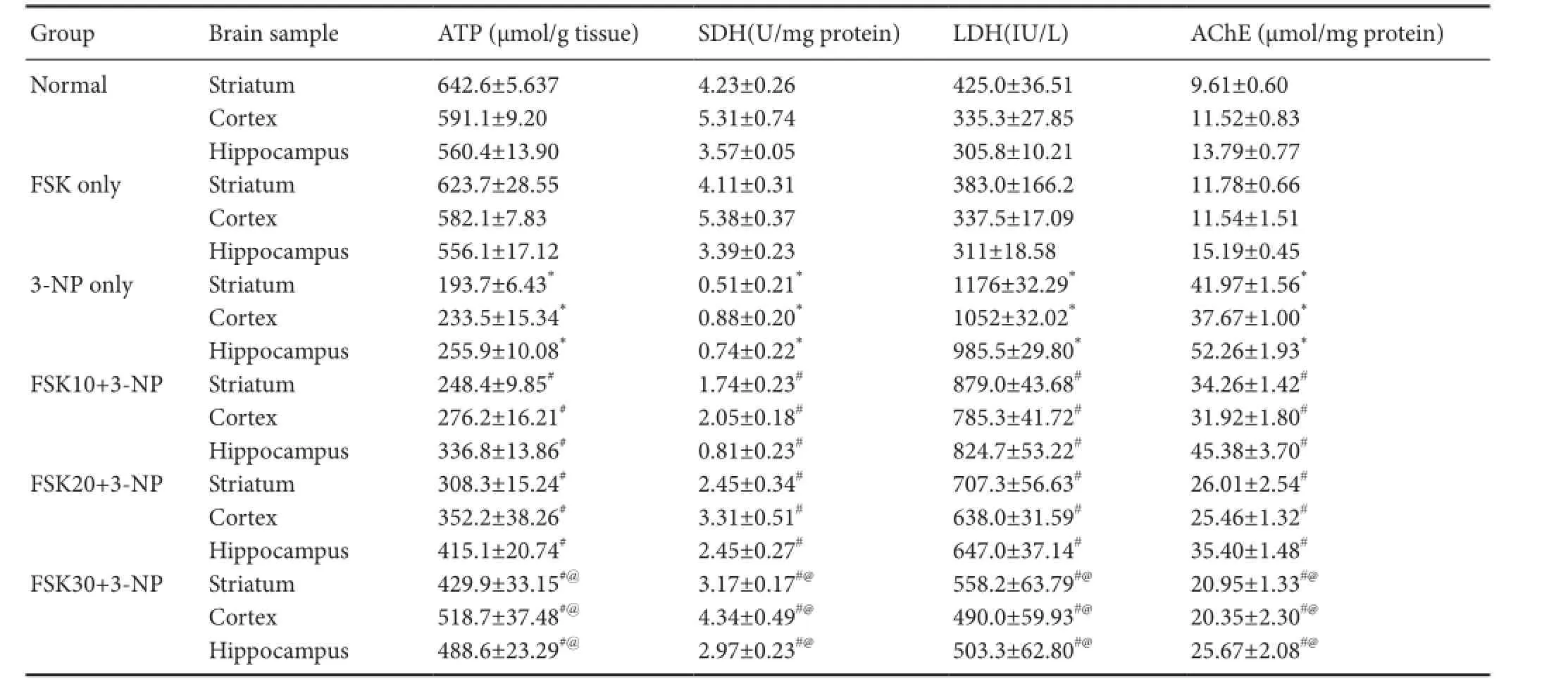
Table 1 Ef f ect of forskolin (FSK) treatment on 3-nitroproprionic acid (3-NP) induced changes in ATP, SDH, LDH and AChE activities in the striatum, cortex and hippocampus of rats
AChE is an enzyme involved in the degradation of synaptic neurotransmitter acetylcholine (Dhir et al., 2008). AChE activity in the homogenate of rat striatum, cortex and hippocampus was signif i cantly increased in the 3-NP only group than in the normal or FSK only group (P <0.05). FSK only injection did not induce obvious alterations in AChE activity when compared to normal rats. AChE activity was signf i ciantly decreased in the FSK10 + 3-NP, FSK20 + 3-NP, and FSK30 + 3-NP groups than in the 3-NP only group (P < 0.05). FSK at 30 mg/kg was more ef f ective in reducing AChE activity than FSK at 10 and 20 mg/kg (P < 0.05; Table 1).
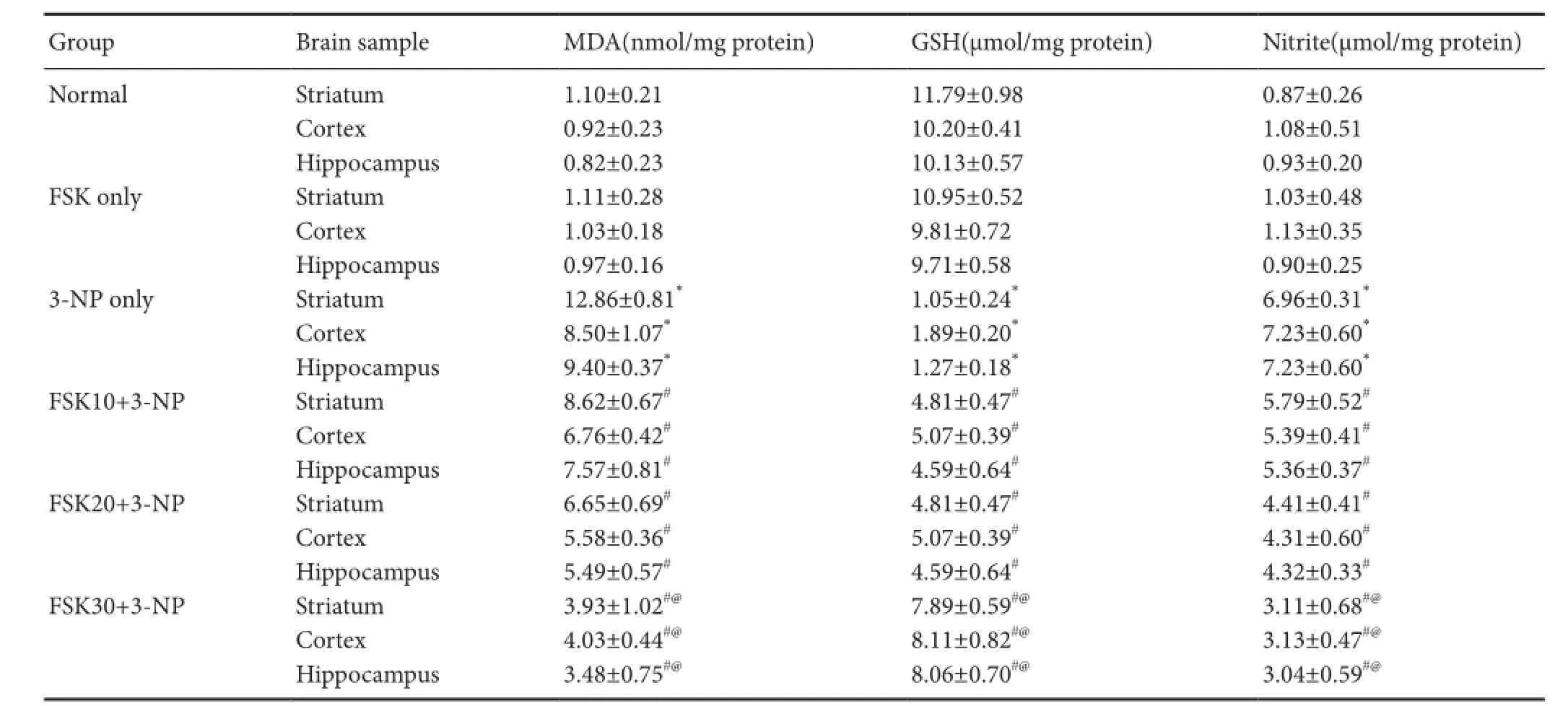
Table 2 Ef f ect of forskolin (FSK) administration on 3-nitroproprionic acid (3-NP) induced changes in MDA, GSH and nitrite levels in the striatum, cortex, and hippocampus of rats
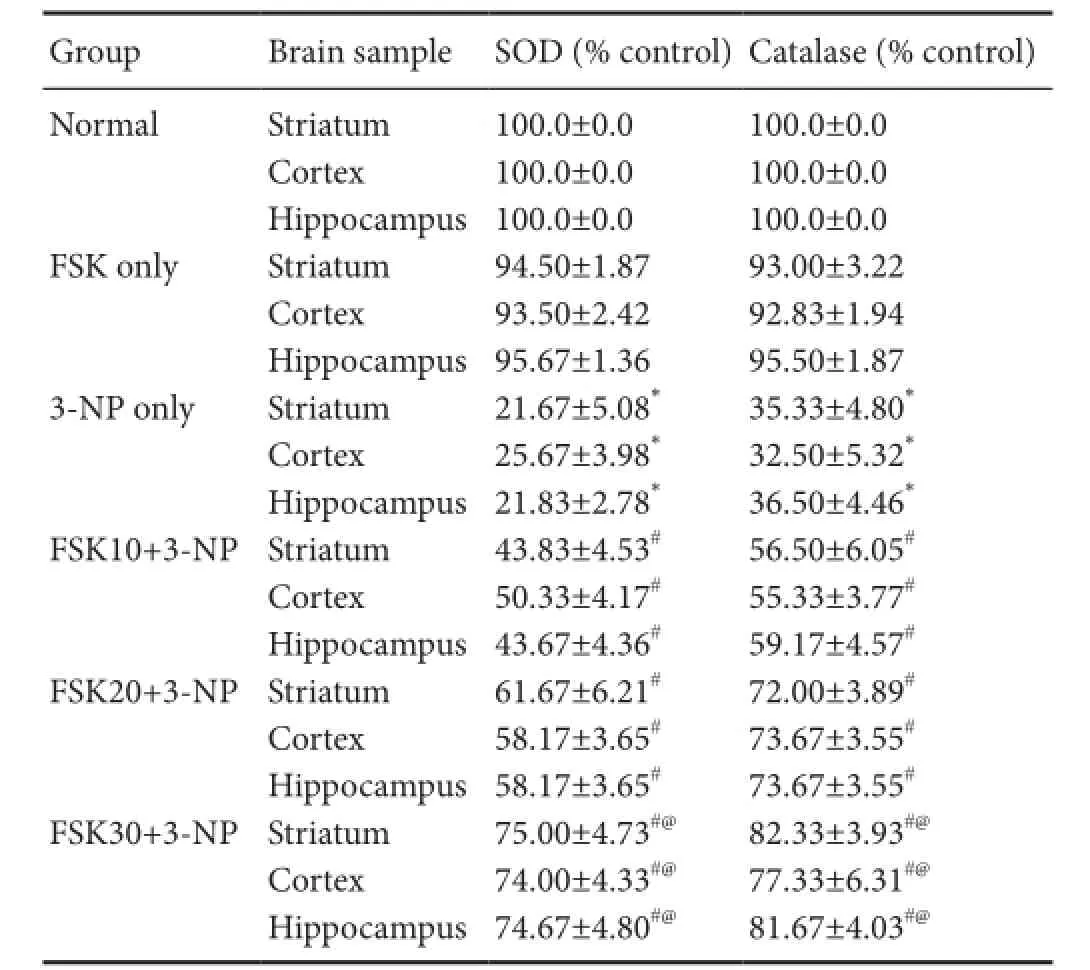
Table 3 Ef f ect of forskolin (FSK) administration on 3-nitroproprionic acid (3-NP) induced changes in SOD and catalase levels in the striatum, cortex, and hippocampus of rats
MDA levels
MDA is an end-product of neuronal cell membrane lipid peroxidation (Dalle-Donnea et al., 2003). MDA level was signif i cantly increased in 3-NP only group than in the normal or FSK only group (P < 0.05). There was no signif i cant difference in MDA level between FSK only and normal groups. MDA level was signif i cantly decreased in the FSK10+ 3-NP, FSK20 + 3-NP, and FSK30 + 3-NP groups than that in the 3-NP only group (P < 0.05). FSK at 30 mk/kg was better effective in lowering MDA levels than FSK at 10 and 20 mg/kg (P < 0.05; Table 2).
GSH levels
GSH is the major antioxidant for reducing free radicals (Liu et al., 2013). GSH level in the brain samples of the striatum, cortex and hippocampus was remarkably decreased in 3-NP-treated rats than in normal or FSK only-treated rats (P < 0.05). There was no obvious difference in GSH level between FSK only and normal groups. FSK 10 and 20 mg/kg greatly increased GSH level (P < 0.05). GSH level was signif icantly increased in FSK30 + 3-NP group than in the FSK10 + 3-NP and FSK20 + 3-NP groups (P < 0.05; Table 2).
Nitrite levels
Nitrite level in the homogenate of rat striatum, cortex and hippocampus was significantly increased in the 3-NP only group thanin the normal or FSK only group (P < 0.05). However, rats treated chronically with FSK 10, 20 and 30 mg/kg resulted in dose-dependent reductions in nitrite levels compared with rats treated with 3-NP (P < 0.05). FSK at 30 mg/kg was more ef f ective in reducing nitrite levels than FSK 10 and 20 mg/kg (P < 0.05; Table 2).
SOD activity
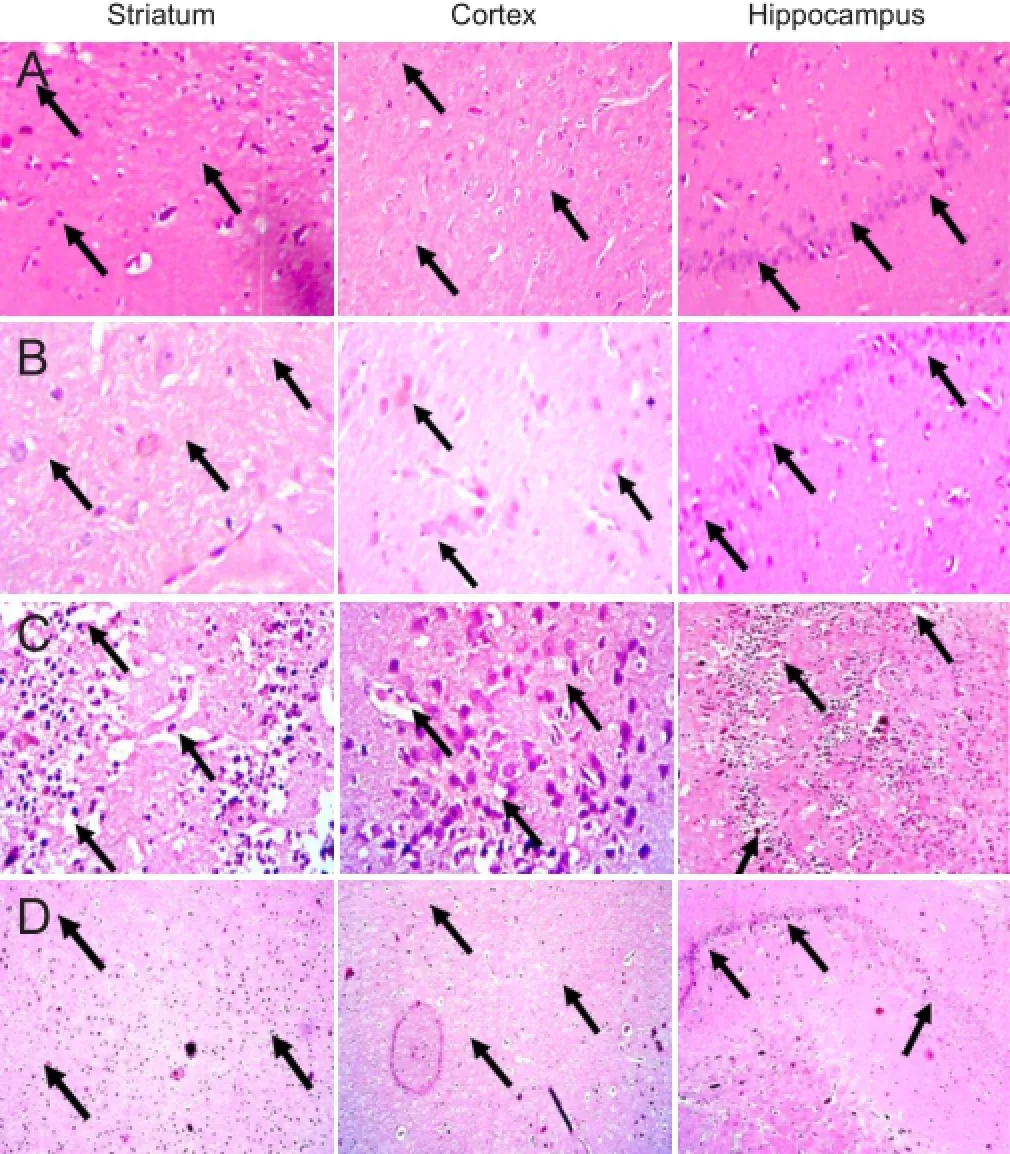
Figure 5 Photomicrographis of hematoxylin-eosin stained brain sections from the striatum, cortex and hippocampus (original magnif i cation × 400).
SOD activity in the homogenate of rat striatum, cortex and hippocampus was signif i cantly decreased in 3-NP only group than in the normal and FSK only groups (P < 0.05). There was no signif i cant dif f erence in alteration between FSK only and normal groups. SOD activity was signif i cantly increased in the FSK10 + 3-NP and FSK20 + 3-NP groups than in the 3-NP only group (P < 0.05). SOD activity was signf i ciantly increased in the FSK30 + 3-NP group than in the 3-NP-only group (P < 0.05). FSK at 30 mg/kg was more ef f ective in reducing SOD activity than FSK 10 and 20 mg/kg (P < 0.05; Table 3).
Catalase activity
Catalase is an enzyme involved in the neurtrilization of oxidized free radicals (Burke et al., 2008). Catalase activity in the homogenate of rat striatum, cortex and hippocampus was signif i cantly increased in 3-NP only group than in the normal and FSK only group (P < 0.05). There was no signif icant dif f erence in alterations in catalase activity between FSK only and normal groups. Catalase activity was signif i cantly increased in the FSK10 + 3-NP and FSK20 + 3-NP groups than in the 3-NP only group (P < 0.05). FSK at 30 mg/kg was more ef f ective in increasing catalase activity than FSK 10 and 20 mg/kg and 3-NP (P < 0.05; Table 3).
Ef f ect of FSK on 3-NP induced histopathological changes in the striatum, cortex and hippocampus of rat brain
Brain sections of the striatum, cortex and hippocampus of rats from the normal (Figure 5A) and FSK only groups (Figure 5B) exhibited shiny cell nuclei and intact cell membrane, indicating that FSK only treatment did not lead to any remarkable ef f ects on histological changes in individual brain sections. On brain sections of the striatum, cortex and hippocampus of rats from the 3-NP only group (Figure 5C), damaged cells with irregular appearance with dense pyknotic nuclei surrounded by marked focal dif f use gliosis compared to that found in the sections from the normal and FSK only groups. Supplementation with FSK 30 mg/kg (Figure 5D) effectivelt attenuated 3-NP induced histological alterations compared to 3-NP only treated rats.
Discussion
There is no cure for Huntington’s disease. However, some phytochemicals and herbal extracts are being investigated to alleviate the symptoms of this disease and prevent it (Thakur et al., 2014). Natural phytochemicals have been shown to be an alterative and treatment strategy to improve neuronal dysfunction in Hungtington’s disease at the optimal level (Nishihara et al., 2003). These natural therapies can reduce oxidative stress and neuronal dysfunction in various brain disorders (Gao et al., 2015). Based on these fi ndings, in this study, we investigated the role of AC activator FSK in the cAMP/CREB activation in 3-NP induced neurodegenerative disorder. 3-NP is a mycotoxin involved in the inhibition of mitochondrial complex II SDH in mitochondrial electron transport chain complex dysfunction and energy failure (Colle et al., 2013) and performs various behavioral and biochemical abnormalities related to memory and motor impairment (Sato et al., 1997; Kim et al., 2000; Cohen and Greenberg, 2008). In this study, rat memory impairment was evaluated in the Morris water maze test. 3-NP intoxicated rats showed remarkable memory acquisition and retention reduction in the Morris water maze test than those untreated or treated with FSK only. 3-NP treatment led to body weight reduction and caused motor and various behavioral dysfunctions like bradykinesia, muscle weakness and rigidity in rats. The present fi ndings are consistent with neuronal dysfunctions mediated behavioral disorders and biochemical changes in 3-NP intoxicated rats as previously reported (Wani et al., 2011; Sundaram et al., 2012; Mehrotra et al., 2015). Body weight was greatly reduced in particular in patients with Huntington’s disease (Harper et al., 2005). Reduced body weight can be considered as an indicator of 3-NP neurotoxicity. At the end stage of Huntington’s disease, patients develop severe motor impairment (Lin et al., 2010). The body weight reduction and motor disorders in 3-NP intoxicated rats can mimic the same dysfunction in patients with Huntington’s disease. In EPM test, transfer latency is an indicator of improving memory impairment (Dhingra and Kumar, 2012). In this study, in EPM test, transfer latency was greatly increased in 3-NP intoxicated rats, showing that the cholinergic system in the brain plays an important role in learning acquisition. Intragastric administration of FSK at 10, 20 and 30 mg/kg exceptionally reduced the transfer latency of 3-NP intoxicated rats. The present findingsrevealed that the brain cholinergic system was involved in the ameliorative effects of FSK on memory impairment of 3-NP-intoxicated rats. 3-NP administration led to loss of grip strength, poor beam walking performance and impaired locomotor function. AC activator FSK greatly ameliorated 3-NP caused memory impairment and motor dysfunction. Reduced levels of cAMP in Huntington’s disease have been documented (Puzzo et al., 2005). However, favorable modulation of cAMP signaling inhibition has been shown to restore cyclic nucleotide pathways in pre-clinical experimental research of Huntington’s disease (Nagakura et al., 2002) and to improve memory impairment and motor dysfunction in rats (Fontan-Lozano et al., 2012). Activated AC/cAMP/PKA/ CREB plays an important role in improving memory impairment and motor dysfunction (Benito and Barco, 2010; Bitner, 2012). In genetic models of Huntington’s disease, CREB plays a multiple role in the coordination and improvement in motor dysfunction (Mehan et al., 2010). CREB level in the striatal region was notably altered during the initial stage of Huntington’s disease caused by 3-NP injection in rats (Choi et al., 2009; Damiano et al., 2010).
Activation of AC has been reported to restore CREB mediated signaling (Nagakura et al., 2002). In the present study, although reduction in ATP level was found to be effective in attenuating 3-NP induced neurotoxicity, the effectiveness of cAMP activation was possibly due to selective and dominant expression of FSK dependent AC enzyme in the striatum, cortex and hippocampus. In the present study, FSK ef ficiently increased ATP level, a sign of cAMP/CREB activation in the brain homogenrate of the striatum, cortex and hippocampus. In the present study, to investigate the neuroprotective mechanism of FSK on brain acetylcholine level, which confirmed the formation of memory and cognitive abilities, AChE level in rat brain homogenate of the striatum, cortex and hippocampus was measured. The hippocampus, amygdale, and cerebral cortex, which are involved in memory and cognitive functioning, are reported to be highly reactive to damage caused by free radicals (Dillingham et al., 1984) and associated with cognitive impairment (Massaad and Klann, 2011). Huntington’s disease is mainly associated with an imbalance between various neurotransmitters in the cholinergic nervous system, leading to memory and cognitive impairments. Loss of cholinergic innervations, as demonstrated by elevated AChE levels, is associated with many neurodegenerative disorders (Oda, 1999). Inhibition of AChE heightened availability of acetylcholine, which is responsible for enhancing cholinergic function (Origlia et al., 2008). In the present study, 3-NP led to an ef f ective in crease in AChE levels, although which has been reported in various studies (Dhir et al., 2008). In the present study, natural phytochemical FSK greatly reduced AChE level. 3-NP intitated energy dysfunction causes excess release of free radicals, which is responsible for the activation of oxidative cascades (Shetty et al., 2015). Oxidative cascades futher activates the reversible secondary cascade, i.e., excitotoxicity (Breton and Rodríguez, 2012). 3-NP has been reported to be involved in the generation of free radicals in the hippocampus, basal ganglia, and cortex in patients with Huntington’s disease, which is associated with memory and cognitive impairments as well as poor motor coordination (Reynolds et al., 1998). However, in case of Huntington’s disease, impaired energy metabolism is mainly due to mitochondrial dysfunction and free radical generation (Monsalve et al., 2012), in which free radical generation mediated cell membrane damage leads to lipid peroxidation. MDA, the end product of lipid peroxidation is considered as an ef f ective target for ameliorating Huntington’s disease-like disorders (Dalle-Donnea et al., 2003). GSH and its free radical redox ameliorating capacity against oxidative stress (Liu et al., 2013), through which various neurodegenerative disorders can be cured, is associated with learning and memory functioning involved in the hippocampus and cortex (Oliveira et al., 2007; Monsalve et al., 2012). SOD and catalase are most powerful anti-oxidant enzymes and play an important role in neutralizing free radicals. Administration of 3-NP resulted in a remarkable reduction in the levels of antioxidant enzymes SOD and catalase (Burke et al., 2008). FSK greatly restored the levels of SOD and catalase. In the present study, 3-NP greatly decreased mitochondrial SDH and GSH activities and increased MDA and nitrite levels. 3-NP intoxicated rats exhibited severe neuronal cell damage as confirmed by increased LDH levels (Tunez et al., 2010). In this study, after 3-NP injection, MDA, nitrite and LDH levels were increased, and SDH and GSH levels were decreased, but FSK administration reversed these alterations, suggesting the anti-oxidative property of FSK. FSK greatly improved mitochondrial dysfunction through elevating ATP levels to keep a balance of neuronal energy. FSK has strong anti-oxidative potential to reduce generation of free radicals.
Results from this study conf i rmed that long term oral FSK perse administration does not exhibit great neuroprotective effects on learning and memory function in normal rats. Oral administration of 10, 20 and 30 mg/kg FSK exhibited notable neuroprotective effects on behavioral paradigms and alteration in biochemical antioxidant enzymes in 3-NP intoxicated rats. This confirms the strong neuroprotective mechanism of FSK in 3-NP intoxicated memory impairment and cognitive dysfunction. The neurotransmitter acetylcholine has been reportedly to play a special role in memory and cognif i ve functions (McGaugh, 2002). Moreover, systemic administration of 3-NP drastically impaired memory retention, resembling Huntington’s disease-like neurodegenerative disorders, and the ameliorative prof i le of natural herbal phytochemica has been explored (Venkatesan et al., 2015). Results from this study demonstrated that FSK reboosted the potent antioxidant prof i le by neutralizing free radicals and conf i rmed the neuroprotective role of FSK in 3-NP intoxicated rats. Further, reduction in AChE levels in this study indicates that FSK exhibits antioxidant capactity against memory impairment. Our results demonstrated motor dysfunction, cognitive impairment and excessive generation of free radicles in animal models of Huntington’s disease caused by 3-NP injection and administration of FSK at 30 mg/kg greatly reversed these changes. This strongly conf i rms the neuroprotectiv ef f ects of FSK on 3-NP-induced neurotoxicity through activating AC.
The cAMP/PKA/CREB pathway reveals a major role in activation of AC by FSK.
Acknowledgments:We express our gratitude to Chairman Dr. Rajender Sra, Secretary Dr. Om Parkash and Counselor Mrs. Ekta Kalra, Rajendra Institute of Technology and Sciences, Sirsa, Haryana, India, for their inspiration and constant support.
Author contributions:SM designed the study and performed the whole surgery; SP worte the paper. SK was responsible for fundraising. All authors approved the fi nal version of this paper.
Conf l icts of interest:None declared.
Aebi H, Wyss, Scherz B, Skvaril F (1974) Heterogeneity of erythrocyte catalase II. Isolation and characterization of normal and variant erythrocyte catalase and their subunits. Eur J Biochem 48:137-45.
Basso M (2012) Targeting Transcriptional Dysregulation in Huntington’s Disease: Description of Therapeutic Approaches. INTECH Open Access Publisher.
Benito E, Barco A (2010) CREB’s control of intrinsic and synaptic plasticity: implications for CREB-dependent memory models. Trends Neurosci 33:230-240.
Binawade Y, Jagtap A (2013) Neuroprotective effect of lutein against 3-nitropropionic acid-induced Huntington’s disease-like symptoms: possible behavioral, biochemical, and cellular alterations. J Med Food 16:934-943.
Bitner RS (2012) Cyclic AMP response element-binding protein (CREB) phosphorylation: A mechanistic marker in the development of memory enhancing Alzheimer’s disease therapeutics. Biochem Pharmacol 83:705—714.
Boulanger L, Poo MM (1999) Gating of BDNF-Induced Synaptic Potentiation by cAMP. Science 284:1982-1984.
Breton RR, Rodríguez JC (2012) Excitotoxicity and oxidative stress in acute ischemic stroke. Stroke 8.
Burke WJ, Kumar VB, Panday N , Pannteon WM, Gan Q, Franko MW, O’Dell M, Li SW, Pan Y, Chung HD, Galvin JE (2008) Aggregation of synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol 115:193-203.
Chaturvedi RK, Beal MF (2013) Mitochondrial diseases of the brain. Free Radical Biol Med 63:1-92.
Chen JY,Wang EA,Cepeda C, Levine MS (2013) Dopamine imbalance in Huntington’s disease: a mechanism for the lack of behavioral fl exibility. Front Neurosci 7:1-14.
Choi EJ, Lee BH (2004) Evidence for genistein mediated cytotoxicity and apoptosis in rat brain. Life Sci 75:499-502.
Choi YS, Lee B, Cho HY (2009) CREB is a key regulator of striatal vulnerability in chemical and genetic models of Huntington’s Disease. Neurobiol Dis 36:259-268.
Chong YH, Shin YJ, Suh YH (2003) Cyclic AMP inhibition of Tumor necrosis factor α production induced by Amyloidogenic C —terminal peptide of Alzheimer’s Amyloidprecursor protein in macrophages: involvement of multiple intracellular pathways and cyclic AMP response element binding protein. Mol Pharmacol 63:690-698.
Cohen S, Greenberg ME (2008) Communication between the synapse and the nucleus in neuronal development, plasticity, and disease. Annu Rev Cell Dev Biol 24:183-209.
Colle D, Santos DB, Moreira EL, Hartwig JM, dos Santos AA, Zimmermann LT, Hort MA, Farina M (2013) Probucol increases striatal glutathione peroxidase activity and protects against 3-nitropropionic acid-induced pro-oxidative damage in rats. PLoS One 8:e67658.
Costa V, Scorrano Z (2012) Shaping the role of mitochondria in the pathogenesis Huntington’s disease. EMBO J 31:1853-1864.
Dalle-Donnea I, Rossib R, Giustarinib D, Milzania A, Colomboa R (2003) Protein carbonyl groups as biomarkers of oxidative stress. Clinica Chimica Acta 329:23-38.
Damiano M, Galvan L, Déglon N, Brouillet E (2010) Mitochondria in Huntington’s disease. Biochim Biophys Acta 1802:52-61.
Delorme T, Najaf i M, Nasr P (2012) The spatial and temporal relationship between oxidative stress and neuronal degeneration in 3-nitropropionic acid model. World J Neurosci 2:234-247.
Deshmukh R, Sharma V, Mehan S, Sharma N, Bedi KL (2009) Amelioration of intracerebroventricular streptozotocin induced cognitive dysfunction and oxidative stress by vinpocetine - a PDE1 inhibitor. Eur J Pharmacol 620:49-56.
Dhingra D, Kumar V (2012) Memory-Enhancing Activity of Palmatine inMice Using Elevated Plus Maze andMorris Water Maze. Adv Pharmacol Sci 1-7.
Dhir A, Akula KK, Kulkarni SK (2008) Tiagabine, a GABA uptake inhibitor, attenuates 3-nitropropionic acid-induced alterations in various behavioral and biochemical parameters in rats. Prog Neuro-Psychopharmacol Biol Psych 32:835—843.
Dillingham MA, Kim JK, Horster MF, Anderson RJ (1984) Forskolin increases osmotic water permeability of rabbit cortical collecting tubule. J Memb Biol 80:243-248.
Ellman G (1959) Tissue sulfhydryl groups. Arch Biochem Biophys 82:70-77.
Ellman GL, Courtney KD, Anders V, Feafterstone RM (1961) A new and rapid colorimetric determination of acetylcholinesterase activity. Biochem Pharmacol 7:88-94.
Fontan-Lozano A, Romero-Granados R, Perez-Villegas EM, Carrión AM (2012) The role of CREB in neuronal plasticity, learning and memory, and in neuropsychiatric disorders. In: Transcription Factors CREB and NF-κB: Involvement in Synaptic Plasticity and Memory Formation (Grilli M, Meneghini V). DOI: 10.2174/97816080525781120101.
Gao Y, Chu SF, Li JP, Zhang Z, Yan JQ, Wen ZL, Xia CY, Mou Z, Wang ZZ, He WB, Guo XF, Wei GN, Chen NH (2015) Protopanaxtriol protects against 3-nitropropionic acid-induced oxidative stress in a rat model of Huntington’s disease. Acta Pharmacol Sin 36:311-322.
Gloerich M, Bos JL (2010) Epac: def i ning a new mechanism for cAMP action. Annu Rev Pharmacol Toxicol 50:355-375.
Gong B, Vitolo OV, Trinchese F, Liu S, Shelanski M, Arancio1 O (2004) Persistent improvement in synaptic and cognitive functions in an Alzheimer mouse model after rolipram treatment. J Clin Invest 114:1624-1634.
Gornall AG, Bardawill CJ, David MM (1949) Determination of serum proteins by means of the biuret reaction. J Biol Chem 177:751—66.
Green LC, Wagner DA, Glogowski J, Skipper PL, Wishnok JS, Tannenbaum SR (1982) Analysis of nitrate, nitrite, and [15N]nitrate in biological fl uids. Anal Biochem 126:131-138.
Harper SQ, Staber PD, He X, Eliason SL, Martins IH, Mao Q, Yang L, Kotin RM, Paulson HL, Davidson BL (2005) RNA interference improves motor and neuropathological abnormalities in a Huntington’s disease mouse model. Proc Natl Acad Sci U S A 102:5820-5825.
Heo H, Yoo M, Han D, Cho Y, Joung I, Kwon YK (2013) Upregulation of TrkB by forskolin facilitated survival of MSC and functional recovery of memory def i cient model rats. Biochem Biophys Res Comm 431:796-801.
Ishrat T, Khan MB, Hoda MN, Yousuf S, Ahmad M, Ansari MA, Ahmad AS, Islam F (2006) Coenzyme Q10 modulates cognitive impairment against intracerebroventricular injection of streptozotocin in rats. Behav Brain Res 171:9-16.
Jadiswami C, Megha HM, Shivsharan BD, Sharanbasappa D, Pandharinath PP, Thippeswamy BS, Veerapur VP (2014) Piroxicam attenuates 3-nitropropionic acid-induced brain oxidative stress and behavioral alteration in mice. Toxicol Mech Methods 24:1-7.
Ji Y, Pang PT,Feng L, Lu B (2005) Cyclic AMP controls BDNF- induced TrkB phosphorylation and dentritic spine formation in mature hippocampal neurons.Nat Neurosci 8:165-172.
Kim GW, Copin JC, Kawase M, Chen SF, Sato S, Gobbel GT, Chan PH (2000) Excitotoxicity is required for induction of oxidative stress and apoptosis in mouse striatum by the mitochondrial toxin, 3-nitropropionic acid. J Cereb Blood Flow Metab 20:119-129.
Kim SH, Thomas CA, André VM, Cummings DM, Cepeda C, Levine MS, Ehrlich ME (2011) Forebrain striatal-specific expression of mutant huntingtin protein in vivo induces cell-autonomous age-dependent alterations in sensitivity to excitotoxicity and mitochondrial function. ASN Neuro 3:e00060.
Kumar P, Kumar A (2009) Protective effect of rivastigmine against 3-nitropropionic acid-induced Huntington’s disease like symptoms: Possible behavioural, biochemical and cellular alterations. Eur J Pharmacol 615:91-101.
Kumar P, Padi SS, Naidu PS, Kumar A (2007) Cyclooxygenase inhibition attenuates 3-nitropropionic acid-induced neurotoxicity in rats: possible antioxidant mechanisms. Fund Clin Pharmacol 21:297-306. Kumar P, Padi SSV, Naidu, PS and Kumar A (2006) Ef f ect of resveratrol on 3-nitropropionic acid- induced biochemical and behavioural changes: possible neuroprotective mechanisms. Behav Pharmacol 17:485-492.
Leenders AG, Sheng ZH (2005) Modulation of neurotransmitter release by the second messenger-activated protein kinases: implications for presynaptic plasticity. Pharmacol Ther 105:69-84.
Lin B, Levy S, Raval AP, Perez-Pinzon MA, DeFazio RA (2010) Forebrain ischemia triggers GABAergic system degeneration in substantial nigra at chronic stages in rats. Cardiovas Psych Neurol 2010:1-16.
Liot G, Bossy B, Lubitz S, Kushnareva Y, Sejbuk N, Bossy-Wetzel E (2009) Complex II inhibition by 3-NP causes mitochondrial fragmentation and neuronal cell death via an NMDA- and ROS-dependent pathway. Cell Death Dif f er 16:899-909.
Liu M, Gua L, Sulkin MS (2013) Mitochondrial dysfunction causing cardiac sodium channel downregulation in cardiomyopathy. J Mol Cell Cardiol 54:25-34.
Maharaj DS, Walker RB, Glass BD, Daya S (2003) 6-Hydroxymelatonin protects against cyanide induced oxidative stress in rat brain homogenates. J Chem Neuroanat 26:103-107.
Massaad CA, Klann E (2011) Reactive oxygen species in the regulation of synaptic plasticity and memory. Antioxid Redox Signal 14:2013-2054.
McGaugh JL (2002) Memory consolidation and the amygdala: a systems perspective. Trends Neurosci 25:456.
Mehan S, Nain VS, Meena H, Sharma D, Sankhla R, Singh M (2010) Comparative ef f ect of phosphodiesterase inhibitors on intracerebroventricular cochicine model of memory impairement in rats. Int J Pharma Prof Res 1:122-136.
Mehrotra A, Kanwal A, Banerjee SK, Sandhir R (2015) Mitochondrial modulators in experimental Huntington’s disease: reversal of mitochondrial dysfunctions and cognitive deficits. Neurobiol Aging 36:2186-2200.
Misra HP, Fridovich I (1972) The role of superoxide anion in the autoxidation of epinephrine and a simple assay for superoxide dismutase. J Biol Chem 247:3170-3175.
Monsalve M, Borniquel S, Valle I, Lamas S (2012) Mitochondrial dysfunction in human pathologies. Front Biosci 12:1131-1153.
Nagakura A, Niimura M, Takeo S (2002) Ef f ects of a phosphodiesterase IV inhibitor rolipram on microsphere embolism-induced defects in memory function and cerebral cyclic AMP signal transduction system in rats. Brit J Pharmacol 135:1783-1793.
Nishihara H, Kizaka-Kondoh S, Insel PA, Eckmann L (2003) Inhibition of apoptosis in normal and transformed intestinal epithelial cells by cAMP through induction of inhibitor of apoptosis protein (IAP)-2. Proc Natl Acad Sci U S A 100:8921-8926.
Oda Y (1999) Choline acetyltransferase: the structure, distribution and pathologic changes in the central nervous system. Pathol Int 49:921-937.
Oliveira JM, Jekabsons MB, Chen S, Lin A, Rego AC, Gonçalves J, Ellerby LM, Nicholls DG (2007) Mitochondrial dysfunction in Huntington’s disease: the bioenergetics of isolated and in situ mitochondria from transgenic mice. J Neurochem 101:241-249.
Origlia N, Kuczewski N, Pesavento E, Aztiria E, Domenici L (2008) The role of cholinergic system in neuronal plasticity: focus on visual cortex and muscarinic receptors. Arch Ital Biol 146:165-188.
Pahan K, Namboodiri AM, Sheikh FG, Smith BT, Singh I (1997) Increasing cAMP attenuates induction of inducible nitric-oxide synthase in rat primary astrocytes. J Biol Chem 272:7786-7791.
Pizzorusso T, Ratto GM, Putignano E, Maf f ei L (2000) Brain-derived neurotrophic factor causes cAMP response element-binding protein phosphorylation in absence of calcium increases in slices and cultured neurons from rat visual cortex. J Neurosci 20:2809-2816.
Puzzo D, Vitolo O, Trinchese F, Jacob JP, Palmeri A, Arancio O (2005) Amyloid-beta peptide inhibits activation of the nitric oxide/cGMP/ cAMP-responsive element-binding protein pathway during hippocampal synaptic plasticity. J Neurosci 25:6887-6897.
Ramanathan M, Babu CS, Justin A, Shanthakumari S (2012) Elucidation of neuroprotective role of endogenous GABAand energy metabolites in middle cerebral artery occluted model in rats. Ind J Exp Biol 50:391-397.
Reynolds DS, Carter RJ, and A. Morton AJ (1998) Dopamine modulates the susceptibility of striatal neurons to 3-nitropropionic acid in the rat model of Huntington’s disease. J Neurosci 18:10116-10127.
Ribeiro FM, Dobransky T, Carvalho EA, Cruz JS, Oliveira FA (2012) Energy Metabolism in Huntington’s Disease. INTECH Open Access Publisher
Rosales-Corral S, Tan DX, Manchester L, Reiter RJ (2015) Diabetes and Alzheimer disease, two overlapping pathologies with the same background: oxidative stress. Oxid Med Cell Longev 2015:985845.
Sassone J, Colciago C, Cislaghi G, Silani V, Ciammola A (2009) Huntington’s disease: The current state of research with peripheral tissues. Exp Neurol 219:385—397.
Sato S, Gobbel GT, Honkaniemi J, Li Y, Kondo T, Murakami K, Sato M, Copin JC, Chan PH (1997) Apoptosis in the striatum of rats following intraperitoneal injection of 3-nitropropionicacid. Brain Res 745: 343-347.
Sharma M, Gupta YK (2003) Ef f ect of alpha lipoic acid on intracerebroventricular streptozotocin model of cognitive impairment in rats. Eur Neuropsychopharmacol 13:241-247.
Shear DA, Dong J, Gundy CD, Haik-Creguer KL, Dunbar GL (1998) Comparison of intrastriatal injections of quinolinic acid and 3-nitropropionic acid for use in animal models of Huntington’s disease. Prog Neuropsychopharmacol Biol Psychiatry 22:1217-1240.
Shetty S, Hariharan A, Shirole T, Jagtap AG (2015) Neuroprotective potential of escitalopram against behavioral, mitochondrial and oxidative dysfunction induced by 3-nitropropionic acid. Ann Neurosci 22:11-18.
Singh S, Jamwal S, Kumar P (2015) Piperine enhances the protective effect of curcumin against 3-NP induced neurotoxicity: possible neurotransmitters modulation mechanism. Neurochem Res 40:1758-1766.
Sundaram RS, Gowtham L, Nayak BS (2012) The role of excitatory neurotransmitter glutamate in brain physiology and pathology. Asian J Pharmaceut Clin Res 5:P1-7.
Thakur T, Mehan S, Kaur R, Kalra S (2014) Huntington’s disease: A hard nut to break. Int J Rec Adv Pharmaceut Res 4:38-58.
Tunez I, Tasset I, Cruz VP, Santamaria A (2010) 3-Nitropropionic acid as a tool to study the mechanisms involved in Huntington’s disease: past, present and future. Molecules 15:878-916.
Venkatesan R, Ji E, Kim SY (2015) Phytochemicals that regulate neurodegenerative disease by targeting neurotrophins: a comprehensive review. Biomed Res Int 2015:814068.
Walker RH (2011) Dif f erential diagnosis of Chorea. Curr Neurol Neurosci Rep 11:385-395.
Wani TA, Al-Omara MA, Zargarb S (2011) Huntington disease: current advances in pathogenesis and recent therapeutic strategies. Int J Pharm Sci Drug Res 3:69-79.
Wills ED (1966) Mechanism of lipid peroxide formation in animal tissue. Biochem J 99:667-676.
Zhang J,Wang Q, Zhu N,Yu M, Shen B, Xiang J, Lin A (2008) Cyclic AMP inhibits JNK activation by CREB-mediated induction of c-FLIPL and MKP-1, thereby antagonizing UV-induced apoptosis. Cell Death Dif f erent 15:1654-1662.
Zhang L, Fang Y, Lian Y, Chen Y, Wu T, Zheng Y, Zong H, Sun L, Zhang R, Wang Z, Xu Y (2015) Brain-derived neurotrophic factor ameliorates learning def i cits in a rat model of Alzheimer’s disease induced by AB1-42. PLoS One 10:1-14.
Zou J, Crews F (2006) CREB and NF-κB transcription factors regulate sensitivity to excitotoxic and oxidative stress induced neuronal cell death. Cell Mol Neurobiol 26:4-6.
Copyedited by Li CH, Song LP, Zhao M
How to cite this article: Mehan S, Parveen S, Kalra S (2017) Adenyl cyclase activator forskolin protects against Huntington’s disease-like neurodegenerative disorders. Neural Regen Res 12(2):290-300.
Open access statement: This is an open access article distributed under the terms of the Creative Commons Attribution-NonCommercial-ShareAlike 3.0 License, which allows others to remix, tweak, and build upon the work non-commercially, as long as the author is credited and the new creations are licensed under the identical terms.
*Correspondence to: Sidharth Mehan, Ph.D., sidh.mehan@gmail.com.
orcid:
0000-0003-0034-835X (Sidharth Mehan)
10.4103/1673-5374.200812
Accepted: 2017-01-16
- 中国神经再生研究(英文版)的其它文章
- Hyperbaric oxygen preconditioning improves postoperative cognitive dysfunction by reducing oxidant stress and inf l ammation
- Nonhuman primate models of focal cerebral ischemia
- The cortical activation pattern during bilateral arm raising movements
- Neuroprotective ef f ect of the Chinese medicine Tiantai No. 1 and its molecular mechanism in the senescence-accelerated mouse prone 8
- Edaravone protects against oxygen-glucose-serum deprivation/restoration-induced apoptosis in spinal cord astrocytes by inhibiting integrated stress response
- Ef f ect of electroacupuncture on the mRNA and protein expression of Rho-A and Rho-associated kinase II in spinal cord injury rats