
散发性包涵体肌炎临床、电生理及病理特点
2017-03-08 09:35:47胡怀强曹霞李靖王树才曹秉振
临床神经病学杂志 2017年1期
胡怀强,曹霞,李靖,王树才,曹秉振
·学术交流·
散发性包涵体肌炎临床、电生理及病理特点
胡怀强,曹霞,李靖,王树才,曹秉振
目的 探讨散发性包涵体肌炎的临床、电生理及病理特点。方法 回顾性分析5例散发性包涵体肌炎患者的临床资料。结果 5例患者均为男性,发病年龄30~54岁,平均43.2岁,出现症状至确诊平均8年。5例患者受累肌肉分布无规律,肌酸激酶正常或轻度增高,EMG呈肌源性损害、部分伴神经源损害或周围神经损害或肌强直电位,肌肉活检5例均有镶边空泡伴肌纤维炎性浸润,1例见不整边红纤维,电镜下3例有管丝包涵体。结论 因无特征性临床表现,散发性包涵体肌炎早期诊断较为困难,其诊断主要依赖于肌肉活检。
散发性包涵体肌炎;病理特点;电生理;临床特点
散发性包涵体肌炎(sIBM)是一种多见于中老年人以缓慢进行性肌无力、肌萎缩为主要临床表现的慢性炎症性肌病。目前我国sIBM少有报道[1-2],发病到确诊至少4年,早期误诊率约50%[3]。为加深临床对sIBM的认识,本文回顾性分析5例sIBM的临床、电生理及病理特点,现报道如下。
1 临床资料
1.1 一般资料 系2000年10月~2015年3月入住济南军区总医院神经内科的经骨骼肌开放活检确诊的sIBM患者5例,均符合2002年中华医学会神经病学分会神经肌肉病学组制定的sIBM诊断标准[4]和2014年Catalán提出的诊断标准[5]。5例患者均为男性;年龄32~59岁,平均51.2岁;发病年龄30~54岁,平均43.2岁;病程2~15年,平均病程8年。1例患者既往有糖尿病病史,5例均无家族遗传病史。
1.2 临床表现 首发症状为双下肢近端肌无力3例,表现为上楼、上坡行走困难及蹲位起立困难;双下肢远端无力伴球麻痹1例;右下肢远端无力1例。2例患者出现疲劳后肌无力症状加重,休息后略减轻。2例病程为15、14年患者就诊时主要表现为四肢无力伴球麻痹,累及眼外肌、眼轮匝肌、面部肌肉、咽喉部肌肉、胸锁乳突肌、颈伸肌、颈屈肌等,临床表现为双眼内收外展受限、眼睑闭合力弱、示齿吹口哨鼓腮不能、构音不清、饮水呛咳、吞咽困难、声音嘶哑、软腭动度减弱、咽反射减弱,抬头力弱;累及斜方肌、胸大肌、三角肌、肱二头肌、股四头肌四肢近端肌肉和双手骨间肌,表现为双上肢近端肌力Ⅲ~Ⅳ级、双下肢近端肌力Ⅱ级,远端重于近端。1例病程5年的患者就诊时累及股四头肌、双手大小鱼际肌,表现为双下肢无力,双上肢近端肌力正常、手握力Ⅴ-级,双下肢近端Ⅱ+级、远端正常。1例病程4年的患者就诊时表现为四肢无力,抬头力弱,四肢近端Ⅲ级、远端正常。1例病程2年的患者就诊时仅累及右下肢远端,表现右下肢远端肌肉萎缩、右足背屈肌力Ⅲ级。4例患者受累肢体肌张力均减低,双侧腱反射(-),深浅感觉查体均正常,病理征阴性。
1.3 实验室检查 5例患者肌酸激酶为168~2480 U/L(正常值26~200 U/L),平均736.00 U/L。4例患者化验血沉、抗O、类风湿因子、甲状腺功能、肿瘤标记物、抗核抗体谱、肌炎抗体谱、血乳酸以及乙肝五项、丙型肝炎抗体、HIV抗体及梅毒螺旋体抗体均正常。2例行腰穿CSF检查示常规、生化均正常。
1.4 EMG 5例患者行常规单极针EMG检查,均见静息时有自发电位,轻收缩为时限缩短、波幅降低、多相波比例增多,重收缩呈早募集,近端肌肉重于远端,呈肌源性损害。1例患者出现双侧胫神经、腓浅神经、腓深神经波幅及传导下降;1例出现肌强直电位;另2例部分肌肉合并神经源性损害。合并糖尿病的患者EMG示左正中神经运动传导速度减慢,双侧尺神经、桡神经感觉传导速度减慢。仅表现右下肢无力的患者EMG见双侧肱二头肌、股四头肌、胫前肌、腓肠肌均呈肌源性损害。
1.5 肌肉病理学检查 5例患者肌肉活检HE染色均可见所检肌肉肌纤维大小不等,萎缩纤维呈角形、不规则形、圆形和椭圆形,部分肌纤维肥大变圆,个别肌纤维出现核内移(图1A)。2例肌纤维核内移明显(图1B);均有散在肌纤维坏死,可见肌内膜下灶性或肌间质内少量炎性细胞浸润,以淋巴细胞和单核细胞浸润为主(图1C),血管周围无明显炎性细胞浸润。4例见到淋巴细胞侵人非坏死性肌纤维内。2例肌间质增宽,间质结缔组织增生明显。5例患者HE、MGT染色均可见散在肌纤维出现镶边空泡(图1A、1B、1D),1例MGT出现典型不整边红纤维(图1E)。5例患者COX染色及Dystrophin-N、C、R端免疫组织化学染色均未见阴性肌纤维,NADH、ORO、PAS染色均为阴性。4例均行肌肉电镜检查,3例肌纤维内可见管丝包涵体(图1F),病程较长的2例均未见到管细丝包涵体。
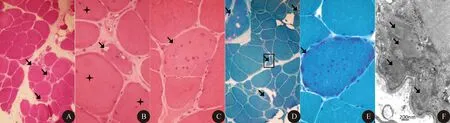
图1 肌肉病理学检查。A:镶边空泡(箭头所示)、肌纤维大小不等、形态变圆、肌间质增宽(HE染色,×200);B:镶边空泡(箭头所示)、肌膜核内移(星号所示)(HE染色,×400);C:炎性细胞浸润非坏死肌纤维(箭头所示)(HE染色,×400);D:镶边空泡(箭头所示)(MGT染色,×200);E:不整边红纤维(箭头所示)(MGT染色,×400);F:电镜可见肌膜下大量管丝包涵体(箭头所示)(×50 000)
2 讨 论
sIBM是一种好发于中老年人以缓慢、进行性肌无力和肌萎缩为主要临床表现的慢性炎性肌肉病,约占炎性肌肉病的30%[6]。Adams等[7]于1965年首次在文献中描述了本病的临床表现,1971年由Yunis等[8]首次命名包涵体肌炎,1978年Carpenter等[9]作为一种独立疾病总结了其临床病理特点。Griggs等[10]于1995年制定了包涵体肌炎的诊断标准,我国于2002年进行了完善性修改[4],2014年Catalán[5]修订了Griggs的sIBM诊断标准。
sIBM起病较晚,多在50岁以后发病,男性多于女性[11],且发病年龄越晚疾病进展越快[12]。本组均为男性,发病年龄30~54岁,平均43.2岁,早于国外报道的发病年龄[3,11]。本组出现症状后平均8年明确诊断,较国外4年确诊时间明显延长[3],一方面与肌肉活检的普及率不高有关,另一方面可能是临床缺乏对该病的认识。本病病因尚不清楚,其发病机制涉及环境因素(如病毒感染)、衰老、基因易感性、自身免疫、毒性蛋白的沉积、肌纤维变性、内质网应激、自噬损害、泛素蛋白酶的破坏、肌生成抑制蛋白的信号调控、线粒体功能异常、核酸代谢异常等[13]。本病为隐匿性起病,缓慢进行性加重,本组患者首先累及双下肢近端(3例)、双下肢远端伴咽喉肌(1例)、右下肢远端(1例),受累肌肉分布无明显规律,四肢近端和远端肌肉在疾病早期均可累及,与sIBM经典表现股四头肌、上肢远端屈肌受累不同[11]。临床以肱二头肌、肱三头肌、前臂肌、髂腰肌、股四头肌和胫前肌受累最常见[14],可呈非对称性[3]。随着疾病进展,本组2例逐渐累及眼外肌、眼轮匝肌、面部肌肉、咽喉肌、胸锁乳突肌、颈伸肌、颈屈肌、斜方肌、胸大肌、三角肌等肌肉,且肢体远端重于近端,这可能与2例病程较长(>14年)有关。部分患者还可累及脊旁肌出现驼背和脊柱侧弯[5]。此外,sIBM较其他炎性肌病更易累及球部肌肉[15]。肌无力和肌萎缩呈比例,病程>2年的4例患者受累肢体肌张力均减低,双侧腱反射均未叩出,需注意与周围神经病的鉴别。2例肌无力变化与疲劳相关,可能存在线粒体功能异常,有待进一步研究。5例均无明显肌痛、肌肉压痛。临床部分sIBM可以合并干燥综合征、系统性红斑狼疮、硬皮病、风湿性关节炎、血小板减少性紫癜等自身免疫性疾病或HIV感染、慢性淋巴细胞性白血病等[11],但本组5例患者无相关临床表现和相关实验室检查未见明显异常。
sIBM患者的肌酸激酶正常或呈轻度增高。研究[2]表明,sIBM早期肌酸激酶明显增高,随着疾病的发展肌酸激酶逐渐降低,病程在5年内的患者的肌酸激酶高于5年病程以上的患者,这可能因受累肌肉病变加重、肌肉萎缩引起。本组4例不超过正常值高限的12倍。1例病程2年的患者肌酸激酶为2480 U/L,病程14、15年的2例患者分别为328 U/L、168 U/L。本组5例患者EMG检查均呈肌源性损害,部分合并神经源性损害、周围神经病变及肌强直。1例临床症状、体征局限于下肢远端的患者,其EMG可见四肢远端、近端均受累,提示EMG可发现亚临床损害的肌肉病变。EMG在sIBM的确诊中无意义[16],但仍是判断肌无力、肌萎缩患者是否存在肌肉病变的关键指标[14]。
随着分子生物学的发展,在sIBM患者中发现了一些分子标记物,但临床确诊仍主要依赖于骨骼肌活检病理学检查[5]。肌肉病理改变可表现为肌纤维肥大、萎缩、坏死、再生、肌膜核内移等形态学改变,以及单核细胞包绕或浸润非坏死肌纤维、镶边空泡、嗜酸性包涵体、淀粉样包涵体、破碎红纤维、COX阴性和SDH阳性肌纤维、肌膜表达组织相容性抗原1和P62、磷酸化tau蛋白、TDP-43等蛋白沉积[11,13],但缺乏特异性[13]。本组5例患者HE、MGT染色均可见散在肌纤维出现镶边空泡,以萎缩肌纤维为主;肌纤维大小不等,见萎缩、肥大、坏死及炎性细胞浸润,血管周围无明显炎细胞;MGT染色示1例可见个别肌纤维肌膜下有不整边红纤维。肌纤维内镶边空泡并非sIBM所特有,还可见于遗传性IBM、有镶边空泡的远端肌病、酸性麦芽糖酶缺陷病、还原体肌病、肌营养不良、慢性酒精性肌病、药物相关性肌病等,因此病理改变有镶边空泡肌纤维时需注意与伴镶边空泡的肌病相鉴别。sIBM电镜下可见管丝包涵体,本组有3例可见管丝包涵体,病程长的2例未见。一方面可能因取材受限致使没有切到存在包涵体的层面,另一方面是否与患者病程较长有关有待进一步研究。因此,在临床上没有必要强调将通过电镜观察到包涵体作为诊断的唯一证据[14]。Griggs等[10]认为,部分肌纤维炎性细胞浸润合并镶边空泡、或合并管丝包涵体是sIBM的特异性改变。
综上所述,sIBM的临床症状、体征、肌酶、EMG均缺乏特异性改变,早期明确诊断困难,确诊主要依赖于肌肉病理改变。对中老年隐匿出现、缓慢进展的肌无力、肌萎缩,需考虑sIBM的可能,应尽早行肌肉活检以明确诊断。
[1]牛丰南,金庆文,张平,等.伴有破碎红纤维的包涵体肌炎的临床及病理学特点(附1例报告)[J].临床神经病学杂志,2013,26:19.
[2]Li K, Pu C, Huang X, et al. Clinicopathologic features of sporadic inclusion body myositis in China[J]. Neurol Neurochir Pol, 2015, 49: 245.
[3]Cortese A, Machado P, Morrow J, et al. Longitudinal observational study of sporadic inclusion body myositis: implications for clinical trials[J]. Neuromuscul Disord, 2013, 23: 404.
[4]中华医学会神经病学分会神经肌肉病学组.包涵体肌炎的诊断标准[J].现代实用医学,2004,16:244.
[5]Catalán M. Selva-O’callaghan a,Grau JM.Diagnosis and classification of sporadic inclusion body myositis(sIBM)[J]. Autoimmun Rev, 2014, 13: 363.
[6]Needham M, Mastaglia FL. Inclusion body myositis: current pathogenetic concepts and diagnostic and therapeutic approaches[J]. The Lancet Neurology, 2007, 6: 620.
[7]Adams RD, Kakulas BA, Samaha FA. A myopathy with cellular inclusions[J]. Trans Am Neurol Assoc, 1965, 90: 213.
[8]Yunis EJ, Samaha FJ. Inclusion body myositis[J]. Lab Invest, 1971, 25: 240.
[9]Carpenter S, Karpati G, Heller I, et al. Inclusion body myositis: a distinct variety of idiopathic inflammatory myopathy[J]. Neurology, 1978, 28: 8.
[10]Griggs RC, Askanas V, Dimauro S, et al. Inclusion body myositis and myopathies[J]. J Neurol Sci, 2002, 199 (Suppl 1):S49.
[11]Mastaglia FL, Needham M. Inclusion body myositis: a review of clinical and genetic aspects, diagnostic criteria and therapeutic approaches[J]. J Clin Neurosci, 2015, 22: 6.
[12]Hori H, Yamashita S, Tawara N, et al. Clinical features of Japanese patients with inclusion body myositis[J]. J Neurol Sci, 2014, 346: 133.
[13]Machado PM, Dimachkie MM, Barohn RJ. Sporadic inclusion body myositis: new insights and potential therapy[J]. Curr Opin Neurol, 2014, 27: 591.
[14]蒲传强.注意散发性包涵体肌炎的诊断[J].中华神经科杂志,2009,42:145.
[15]Lynn SJ, Sawyers SM, Moller PW, et al. Adult-onset inflammatory myopathy: North Canterbury experience 1989-2001[J]. Intern Med J, 2005, 35: 170.
[16]Rose MR, ENMC IBM Working Group. 188th ENMC international workshop: inclusion body myositis, 2-4 December 2011, naarden, the Netherlands[J]. Neuromuscul Disord, 2013, 23: 1044.
Clinical, electrophysiological and myopathological features of sporadic inclusion body myositis
HUHuai-qiang,CAOXia,LIJing,etal.
DepartmentofNeurology,GeneralHospitalofJinanMilitaryCommand,Jinan250031,China
Objective To investigate the clinical, electrophysiological and myopathological features of sporadic inclusion body myositis (sIBM).Methods The clinical data of 5 sIBM patients were retrospectively analyzed. Results Five cases were all male. The onset of symptoms varied from 30 to 54 years old with an average age of 43.2 years old. The average times from onset to definite diagnose were 8 years. The affected muscles of 5 cases with sIBM distributed randomly. The creatine kinase values were normal or mild elevated in the five cases. EMG showed myogenic changes in the all cases, and neurogenic changes or peripheral neuropathy or rigidity electric potential in individual case. Rimmed vacuoles and endomysial inflammation were found in all of the patients. Ragged red fibers appears in one of them. Electron microscope showed tubulofilamentous inclusions in the 3 cases. Conclusion It’s difficult to definitely diagnose on the initial stage of sIBM because of the lack of special clinical manifestations, and the definite diagnose primarily relies on muscle biopsy.
sporadic inclusion body myositis;pathological feature;electrophysiology;clinical feature
250031中国人民解放军济南军区总医院神经内科
曹秉振
R746
A
1004-1648(2017)01-0058-03
2015-12-09
2016-03-08)
猜你喜欢
中国临床医学影像杂志(2022年6期)2022-07-26 07:17:24
中国临床医学影像杂志(2022年5期)2022-07-26 07:11:54
数字海洋与水下攻防(2021年2期)2021-05-08 08:01:26
国际放射医学核医学杂志(2021年10期)2021-02-28 08:43:54
天津农学院学报(2016年2期)2016-12-01 05:40:05
系统工程与电子技术(2016年2期)2016-04-16 05:17:00
黄河之声(2016年22期)2016-02-03 07:59:25
船海工程(2015年4期)2016-01-05 15:53:28
计算物理(2014年1期)2014-03-11 17:00:22
海南师范大学学报(自然科学版)(2012年3期)2012-09-11 00:46:12
