
长骨炎症性肌纤维母细胞性肿瘤的临床病理观察
2017-03-07 23:00:35宫丽华刘巍峰孙晓淇黄啸原
临床与实验病理学杂志 2017年5期
宫丽华,刘巍峰,孙晓淇,张 铭,丁 宜,黄啸原
长骨炎症性肌纤维母细胞性肿瘤的临床病理观察
宫丽华1,刘巍峰2,孙晓淇1,张 铭1,丁 宜1,黄啸原1
目的 探讨骨内炎症性肌纤维母细胞性肿瘤(inflammatory myofibroblastic tumor, IMT)的临床病理特征、免疫表型、诊断及鉴别诊断。方法 采用HE、免疫组化EnVision两步法染色对4例骨内IMT的临床病理学特征进行分析并复习相关文献。结果 4例IMT中女性1例,男性3例,发生于胫骨、股骨各2例,其中3例同时累及骨及软组织。组织学上由肌纤维母细胞样梭形细胞增生伴浆细胞、淋巴细胞、嗜酸性粒细胞等炎细胞浸润。免疫表型:vimentin、SMA、actin、H-caldesmon、CD34均呈阳性,部分细胞表达CD68。结论 IMT是具有局部侵袭性的交界性肿瘤,临床较罕见,应与一些良性及恶性的软组织肿瘤及肿瘤样病变鉴别。
炎症性肌纤维母细胞性肿瘤;骨;免疫表型;诊断;鉴别
炎症性肌纤维母细胞性肿瘤(inflammatory myofibroblastic tumor, IMT)是一种少见的良性但具有局部侵袭性的肿瘤,组织学上由肌纤维母细胞样和纤维母细胞样的梭形细胞组成,伴浆细胞、淋巴细胞、中性粒细胞浸润[1]。最初由Brunn于1939年描述2例发生于肺的良性梭形细胞肿瘤,从而引起关注[2]。1990年,Pettinato等[3]首先报道20例发生于肺的IMT,继而有报道认为除肺外也可以发生于其他器官,如儿童及年轻人的腹部软组织及内脏,但很少累及肢端[4];发生于骨内时主要见于颞骨[5]、下颌骨[6],累及长骨罕见。本文现报道4例发生于长骨的IMT,其中3例同时累及骨及软组织,探讨其临床及组织学特征并结合文献对其临床特点、病理学特征、细胞遗传学表现及预后进行分析,以提高对IMT的认识水平。
1 材料与方法
1.1 临床资料 例1,男性,25岁。左大腿疼痛3年,加重1年伴肿胀3个月。3个月前曾于外院活检并化疗。我院骨肿瘤科查体:左大腿下端外侧可见陈旧性手术瘢痕,可触及包块,质硬,边界不清,无活动,有压痛。CT可见股骨远端后侧溶骨破坏,边界清楚,皮质破坏,后侧可见软组织包块(图1)。MRI:左腘窝上方外侧可及软组织肿块,股骨受压(表1)。手术行股骨下段瘤段截除术。
例2,女性,51岁。右膝疼痛行走不适4年,2个月前症状加重明显。我院行穿刺活检,病理提示:梭形细胞呈浸润性生长,考虑间叶性肿瘤(FNCLCC I级)。骨科查体:右小腿近端肿胀,右下肢内翻畸形。X线检查可见胫骨干中上段骨破坏,基质不均匀,边界不清楚,皮质有破坏,有骨膜反应(图2)。CT示软组织窗和软组织加强窗可见胫骨近端肿瘤,皮质破坏,并形成软组织包块,增强后可见明显强化影像(表1)。手术行右胫骨上段瘤段截除术。
例3,男性,49岁。右小腿间断性疼痛6个月。CT示右胫骨病变,MRI示右胫骨中下段髓腔内可见铸型状软组织密度影,密度均匀,轻度强化(图3)。穿刺活检病理考虑梭形细胞病变,性质难以确定(表1)。入院行胫骨骨干瘤段截除术。
例4,男性,38岁。右大腿下段疼痛不适1个月。1个月前无明显诱因感右大腿下段疼痛,未诊治。2周前触及右大腿内侧肿块,逐渐增大。外院活检组织经我院病理会诊为IMT。骨科查体:右大腿下端内侧可触及一包块,质硬,边界不清,无活动,有压痛。X线示右大腿下段溶骨性破坏。PET-CT发现右股骨上段及右髋臼摄取增高(图4,表1)。我院行右髋臼穿刺活检术。因本例患者为多发病变,未行进一步治疗。
1.2 方法 4例手术标本均经10%中性福尔马林固定,石蜡包埋,3 μm厚连续切片并常规行HE染色。免疫组化采用EnVision两步法,所用一抗vimentin、CK、SMA、H-caldesmon、actin、S-100、ALK、CD31、CD34、CD68、CD3、CD20、CD30分别购自Leica、DAKO、Zymed、北京中杉金桥、福州迈新公司,二抗购自丹麦DAKO公司。

表1 4例长骨IMT的临床资料
2 结果
2.1 眼观 例1送检股骨下段标本,长16 cm,最大径11 cm,切面骨皮质外见灰黄色肿瘤组织,大小9 cm×7 cm×7 cm,切面灰白、灰黄色,质软,鱼肉状,伴囊变,局灶见骨皮质破坏,髓腔内可见肿瘤。例2送检组织分别为骨内病变及骨外病变,骨内病变:灰白色破碎组织一堆,总直径8 cm,切面灰白色,实性,质脆。骨外病变:灰白色囊壁样破碎组织一堆,大小6 cm×5 cm×3 cm,切面灰白色,实性,质脆。例3送检右胫骨切除标本,长14.5 cm,最大径3.5 cm,距一端4.5 cm,另一端3 cm,髓腔内见灰黄色组织,大小2 cm×0.8 cm×0.5 cm,质韧。例4活检标本:送检碎组织一堆,总体积1 cm×1 cm×0.8 cm,灰白、灰褐色,质软。
2.2 镜检 由增生的梭形纤维母细胞/肌纤维母细胞组成,间质伴大量炎细胞浸润,肿瘤周边部呈纤维瘤病样形态,可见破坏骨组织。例1炎细胞增生显著伴弥漫分布,以淋巴细胞为主,可见肥大细胞夹杂其间。瘤细胞散在分布于炎细胞间,呈胖梭形,胞质嗜酸性,少部分细胞核梭形,大部分细胞核圆形、卵圆形,可见小核仁,未见核分裂象(图5)。部分细胞胞质丰富、嗜酸性,呈横纹肌样细胞形态。间质小血管及血管内皮细胞增生活跃。例2、例4炎细胞呈灶状分布,伴淋巴滤泡形成(图6),以淋巴细胞及浆细胞为主。梭形的肿瘤细胞呈束状分布,细胞伸长状,胞质红染,核长梭形,杆状,两端钝圆。部分区域细胞稀疏,胶原化显著,似促结缔组织增生性纤维瘤样形态并伴骨破坏。例3形态相似,肿瘤细胞呈显著的Storiform排列,核圆形,伴胶原纤维形成,形似纤维组织细胞瘤样结构(图7)。局灶见散在钙化及宿主骨的破坏。炎细胞小簇状聚集,分布于瘤组织间。近肿瘤边缘区可见反应性新骨形成。
2.3 免疫表型 肿瘤细胞弥漫性表达vimentin、SMA(图8)、actin、H-caldesmon、CD34,部分细胞表达CD68。其余标志物均阴性。
2.4 随访 4例术后随访11~29个月,未见肿瘤复发及转移。
3 讨论
关于IMT的性质一直存在争议。Coffin等[1]认为其是一种良性非转移性病变,但存在复发及持续局部生长的潜能。WHO(2013)骨及软组织肿瘤将其定义为由肌纤维母细胞样及纤维母细胞样梭形细胞构成,伴炎细胞(浆细胞、淋巴细胞及中性粒细胞)浸润,并归类为交界性肿瘤[7]。其同义词包括:浆细胞肉芽肿、炎性假瘤、炎性肌纤维细胞增生和炎性纤维肉瘤等[8]。
3.1 临床特点 目前,IMT病因学尚不清楚,根据术后、创伤后及感染后的病例报道推测该病变可能起初为反应性,但最终会转化为真性肿瘤[4]。过敏性、免疫性及感染性机制均有可能,EB病毒感染也与40%的IMT有关[9]。肺外IMT常发生于儿童及年轻人,平均年龄10岁,但也可能发生于80岁,年纪较大的患者诊断时应格外谨慎。男女发病比为1 ∶1.4。最常见的临床表现为无意发现的胸腹部包块,少部分病例(15%~30%)可伴难以解释的发热、体重减轻、贫血、血沉加快、高丙球蛋白血症,症状可持续几周甚至几个月[4]。肺为最常见的受累部位,肺外器官包括胃、肠系膜、网膜、肾、肝、脾、食管及淋巴结,头颈、躯干及肢端为少见部位[3]。发生于长骨的患者仅见5例报道(表2),年龄分别为54、18、27、40、41岁,分别发生于肱骨(1例)、胫骨(1例)与股骨(3例),其中2例伴软组织侵犯[10-12]。本组3例与上述5例相似,发病年龄偏大,胫骨2例,股骨2例,其中3例伴软组织侵犯。IMT的影像学并无特异性,X线显示透光性、非膨胀性的界限不清或较清的病变,无硬化缘及骨膜反应。核磁共振显示T1低信号T2高信号的不均一成像,增强后,依据纤维成分的多少及细胞丰富程度不等显示轻~中度强化。

表2 文献报道5例长骨IMT的临床资料
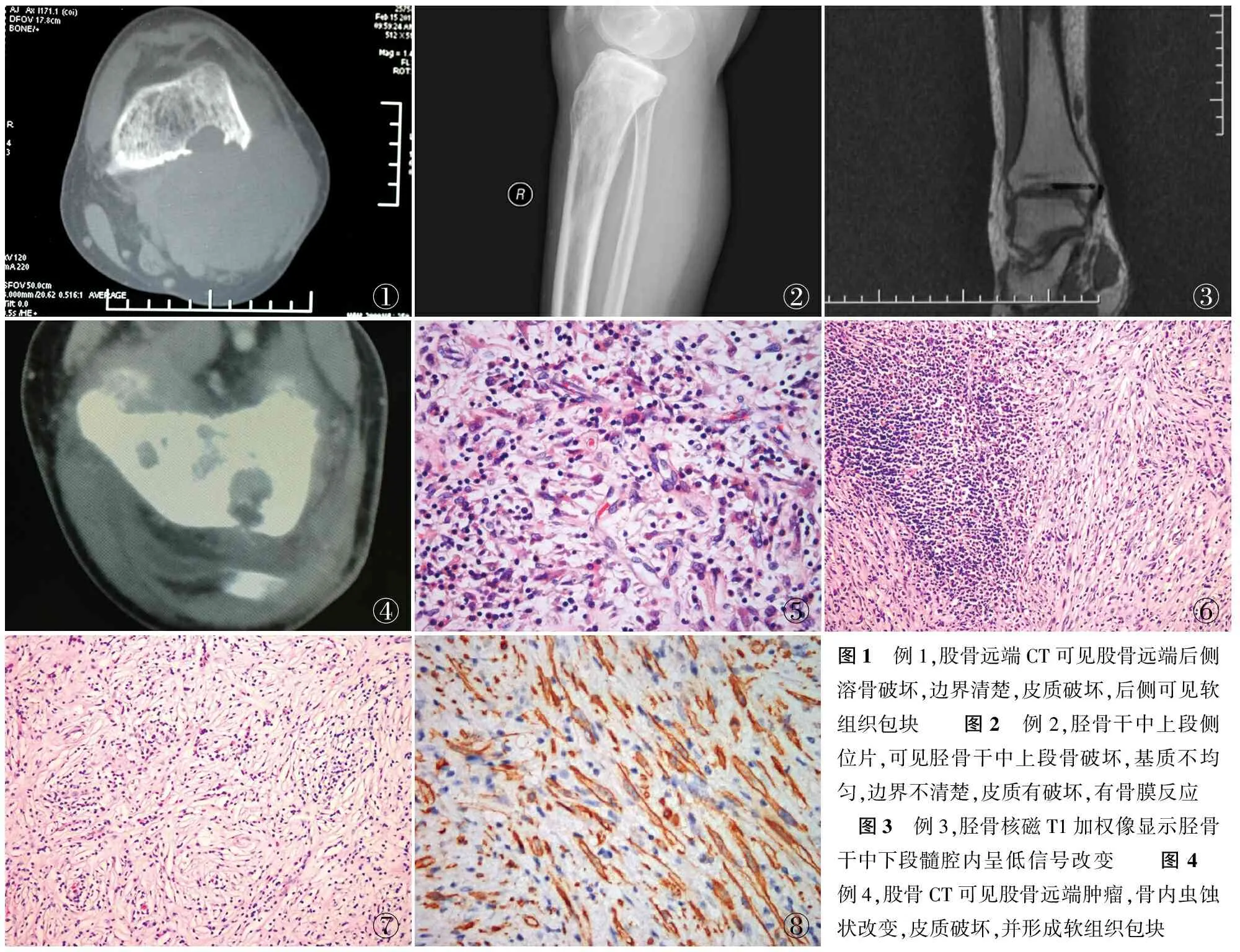
①②③④⑤⑥⑦⑧图1 例1,股骨远端CT可见股骨远端后侧溶骨破坏,边界清楚,皮质破坏,后侧可见软组织包块 图2 例2,胫骨干中上段侧位片,可见胫骨干中上段骨破坏,基质不均匀,边界不清楚,皮质有破坏,有骨膜反应 图3 例3,胫骨核磁T1加权像显示胫骨干中下段髓腔内呈低信号改变 图4 例4,股骨CT可见股骨远端肿瘤,骨内虫蚀状改变,皮质破坏,并形成软组织包块
图5 瘤细胞散在分布于炎细胞间,呈胖梭形,胞质嗜酸性,核圆形、卵圆形,可见小核仁 图6 炎细胞灶状聚集,伴淋巴滤泡形成 图7 梭形的肿瘤细胞呈显著的Storiform排列 图8 梭形细胞表达SMA,EnVision两步法
3.2 组织学特征 IMT肿瘤直径2~20 cm,平均6 cm。常为实性,切面灰白色或褐色,质较韧或橡胶样,偶尔质软胶冻样。少数出血、坏死或钙化[13]。IMT镜下可有3种形态成分,第1种形态:疏松排列的星形、胖梭形细胞分布于水肿、黏液样间质内,小血管形成不规则的网,伴炎细胞浸润,形成肉芽组织样或结节性筋膜炎样结构。间质细胞核空泡状,胞质丰富、嗜酸性,形似横纹肌母细胞。可见核分裂象,但无病理性核分裂。浸润的炎细胞包括淋巴细胞、中性粒细胞及嗜酸性粒细胞。在结节性筋膜炎样结构中,嗜酸性粒细胞而非浆细胞占主要地位。第2种形态:显示致密的梭形细胞增生,呈束状排列或Storiform排列,可见细胞密集区及稀疏区。梭形细胞核伸长,两端钝圆。浆细胞常占主导地位,可聚集呈灶状或散在分布。可伴淋巴细胞增生并形成反应性滤泡中心。如缺少炎细胞的浸润,梭形细胞区域可能与肌源性或纤维组织源性肿瘤相似。如果胶原丰富,则类似于纤维瘤病或肌纤维瘤病。第3种形态:形成瘢痕样或韧带样型纤维瘤病样形态,细胞增生不活跃,浆细胞或淋巴细胞陷于致密的嗜伊红的基质内。镜下常3种成分混合,在同一例中一种或两种占优势。少部分病例可见不规则钙化或骨化,炎性背景是其显著的组织形态特征,通过广泛细致的取材可见到上述3种组织特征。Coffin对比复发前后的组织形态发现,复发后细胞高度非典型性,核空泡状,核仁明显,可见病理性核分裂象,可能提示组织形态转化为组织细胞源性[1]。Spencer报道27例肺IMT中2例发生肉瘤样转化,但由于缺乏随访信息,其临床意义难以评价。
鉴于其组织形态的复杂性,不建议行穿刺活检诊断。本组4例均在术前行穿刺活检,1例结合外院活检组织给予明确诊断,2例穿刺报告梭形细胞肿瘤,其中1例性质难以确定,1例根据穿刺的少量组织给予区域性FNCLCC分级评估。1例穿刺报告血管源性肿瘤,与术后病理差异较大。
3.3 免疫表型及细胞遗传学 肿瘤细胞表达vimentin、actin、desmin、CD34、CD68,36% IMT的表达CK,不表达Myoglobin。细胞遗传学显示约50%的软组织IMT存在2号染色体短臂ALK受体酪氨酸酶的基因重排。因此,ALK是IMT的敏感性标志物,但ALK阴性的IMT形态与ALK阳性的无法鉴别[14]。40%~60%的病例可显示ALK阳性[15],本组4例皆为ALK阴性。
3.4 鉴别诊断 本组4例均发生于长骨内,故需与骨内的肿瘤鉴别。(1)平滑肌肉瘤:绝大部分发生在软组织,瘤细胞丰富,核雪茄状,异型性明显,可见病理性核分裂象。少见浆细胞和淋巴细胞浸润。(2)炎症性恶性纤维组织细胞瘤:又称未分化多形性肉瘤,多见于成人,细胞高度异型性及多形性明显,核分裂象多见,并见病理性核分裂象。炎细胞浸润明显,局灶可以显示相似的炎性肌纤维母细胞肿瘤样形态。(3)骨的促结缔组织增生性纤维瘤:由增生的梭形纤维母细胞和胶原纤维组成,缺乏异型性,核分裂象少见,呈浸润性生长。免疫组化标记β-catenin胞质阳性。
3.5 治疗及预后 IMT有局部侵袭性,治疗主要以外科完整切除;化疗有时适用于多灶或复发的病变,放疗效果不确定。有文献报道[16]血管侵犯以及肉瘤样转化。IMT发生于肺的复发率可能<2%,肺外器官可达25%[17],且复发与孤立性病灶的不完整切除有关,多在术后1年复发,远处转移少见(<5%)[18]。
[1] Coffin C M, Watterson J, Priest J R,etal. Extrapulmonary inflammatory myofibroblastic tumor (inflammatory pseudotumor). A clinicopathologic and immunohistochemical study of 84 cases [J]. Am J Surg Pathol, 1995,19(8):859-872.
[2] Brunn H. Two interesting benign lung tumors of contradictory histopathology: remarks on the necessity for maintaining chest tumor registry[J]. J Thorac Surg, 1939,9(1):119-131.
[3] Pettinato G, Manivel J C, De Rosa N,etal. Inflammatory myofibroblastic tumor (plasma cell granuloma): clinicopathologic study of 20 cases with immunohistochemical and ultrastructural observations[J]. Am J Clin Pathol, 1990,94(5):538-546.
[4] Coffin C M, Humphrey P A, Dehner L P. Extrapulmonary inflammatory myofibroblastic tumor: a clinical and pathological survey[J]. Semin Diagn Pathol, 1998,15(2):85-101.
[5] Gawande P D, Sambhus M, Garde J B,etal. Aggressive inflammatory pseudotumor of the mandible[J]. J Craniofac Surg, 2012,23(4):1101-1103.
[6] Cho Y S, Kim S M, Chung W H,etal. Inflammatory pseudotumour involving the skull base and cervical spine[J]. J Laryngol, 2001,115(1):580-584.
[7] Fletcher C D, Bridge J A, Hogendoorn P C. WHO classification of tumors of soft tissue and bone[M]. Lyon: IARC Press, 2013:83-85.
[8] Savvidou O D, Sakellariou V I, Papakonstantinou O,etal. Inflammatory myofibroblastic tumor of the thigh: presentation of a rare case and review of the literature[J]. Case Rep Orthop, 2015,2015:814241.
[9] Arber D A, Weiss L M, Chang K L. Detection of Epstein-Barr virus in inflammatory pseudotumor[J]. Semin Diagn Pathol, 1998,15(2):155-160.
[10] Baker J F, Lui D F, Cavanagh M, Hurson B J. Inflammatory myofibroblastic tumour of the tibia[J]. Joint Bone Spine, 2010,77(5):488-489.
[11] Sciot R, Dal Cin P, Fletcher C D,etal. Inflammatory myofibroblastic tumor of bone: report of two cases with evidence of clonal chromosomal changes[J]. Am J Surg Pathol, 1997,21(10):1166-1172.
[12] Chen J, Li H, Yang Z,etal. Inflammatory myofibroblastic tumor of bone: two cases occurring in long bone[J]. Skeletal Radiol, 2011,40(1):117-122.
[13] Meis J M, Enzinger F M. Inflammatory fibrosarcoma of the mesentery and retroperitoneum. A tumor closely simulating inflammatory pseudotumor[J]. Am J Surg Pathol, 1991,15(12):1146-1156.
[14] Dehner L P. Inflammatory myofibroblastic tumor: the continued definition of one type of so-called inflammatory pseudotumor[J]. Am J Surg Pathol, 2004,28(12):1652-1654.
[15] Coffin C M, Hornick J L, Fletcher C D. Inflammatory myofibroblastic tumor: comparison of clinicopathologic, histologic, and immunohistochemical features including ALK expression in atypical and aggressive cases[J]. Am J Surg Pathol, 2007,31(4):509-520.
[16] Coffin C M, Patel A, Perkins S,etal. ALK1 and p80 expression and chromosomal rearrangements involving 2p23 in inflammatory myofibroblastic tumor[J]. Mod Pathol, 2001,14(6):569-576.
[17] Janik J S, Janik J P, Lovell M A,etal. Recurrent inflammatory pseudotumors in children[J]. J Pediatr Surg, 2003,38(10):1491-1495.
[18] 雷伟华,邹绮嫦,郭锦辉,等. 炎性肌纤维母细胞肿瘤11例临床病理分析[J]. 临床与实验病理学杂志, 2015,31(12):1422-1425.
Inflammatory myofibroblastic tumor of long bone:a clinicopathologic analysis and review of literatures
GONG Li-hua1, LIU Wei-feng2, SUN Xiao-qi1, ZHANG Ming1, Ding Yi1, HUANG Xiao-yuan1
(1DepartmentofPathology,2DepartmentofOrthopedicOncology,BeijingJishuitanHospital,Beijing100035,China)
Purpose To study the clinicopathologic features of inflammatory myofibroblastic tumor (IMT) of long bone. Methods HE and immunohistochemistry of EnVision two-step were used to observe the clinical, radiological, histological and immunophenotype features of IMT of bone. The literatures were reviewed. Results 4 cases of IMT of bone were respectively located in the tibia (2 cases) and femur (2 cases). Histologically, the lesions were characterized by collagen-rich and spindled to plump myofibroblast-like cells and a variable admixture of inflammatory cells. Immunohistochemical study showed that the vimentin, SMA, actin, H-caldesmon and CD34 were positive. Conclusion The IMT is a rare and locally aggressive tumor. The diagnosis should combine the histological characters with immunohistochemical results and should be differentiated from the other tumors and tumor-like lesions.
inflammatory myofibroblastic neoplasm;bone;immunophenotypes;diagnosis;differentiate
北京积水潭医院1病理科、2骨肿瘤科,北京 100035
宫丽华,女,博士,副主任医师。Tel:(010)58516823, E-mail: lhgong2005@126.com
时间:2017-5-17 23:53 网络出版地址:http://kns.cnki.net/kcms/detail/34.1073.R.20170517.2352.014.html
R 739.9
A
1001-7399(2017)05-0534-05
10.13315/j.cnki.cjcep.2017.05.014
接受日期:2017-03-07
猜你喜欢
首都食品与医药(2023年14期)2023-07-17 07:04:52
中国药学药品知识仓库(2022年8期)2022-05-09 13:54:24
医药前沿(2018年13期)2018-04-20 11:29:04
西安建筑科技大学学报(自然科学版)(2016年5期)2016-11-10 02:39:26
中国卫生标准管理(2015年16期)2016-01-20 09:26:16
哈尔滨医药(2015年5期)2015-12-01 03:58:08
实用手外科杂志(2015年3期)2015-08-27 01:53:06
安徽医科大学学报(2015年11期)2015-07-20 02:41:08
中国医药科学(2015年6期)2015-07-18 01:30:09
中国实用医药(2013年12期)2013-02-02 19:00:32
