
阿法替尼的合成工艺改进
2017-02-24 11:21李文倩宋国强万屹东高有军冯筱晴
合成化学 2017年2期
李文倩, 宋国强, 万屹东, 高有军, 冯筱晴,3*
(1. 常州大学 制药与生命科学学院,江苏 常州 213164; 2. 常茂生物化学工程股份有限公司,江苏 常州 213164; 3. 南京工业大学 化工学院,江苏 南京 210009)
·制药技术·
阿法替尼的合成工艺改进
李文倩1, 宋国强1, 万屹东2, 高有军2, 冯筱晴2,3*
(1. 常州大学 制药与生命科学学院,江苏 常州 213164; 2. 常茂生物化学工程股份有限公司,江苏 常州 213164; 3. 南京工业大学 化工学院,江苏 南京 210009)
以2-氨基-4-氯苯甲酸为原料,经环合、硝化、氯代和胺化后,采用一锅两步法制得关键中间体4-[(3-氯-4-氟苯基)氨基]-6-硝基-7-[(S)-四氢呋喃-3-基氧基]-喹唑啉(4); 4依次经还原、酰胺化、HWE反应合成阿法替尼,总收率55.7%,含量98%,其结构经1H NMR和LC-MS确证。
阿法替尼; 药物合成; 一锅法; 工艺改进
目前,1的合成已有较多报道[3-12],其主要合成路线有:(1)以2-氨基-4-氯苯甲酸(9)为原料,经环合、硝化、氯代、胺化、磺酰化和醚化等8步反应合成1[7,13],总收率28%。该路线存在溶剂复杂,反应路线较长,收率偏低等缺点。(2)以2-氨基-4-氟苯甲酸为原料,经环合、硝化、氯代、胺化和醚化等7步反应合成1,总收率27%[12]。方法(2)虽然步骤减少一步,但原料价格昂贵,成本较高。因此,改进1的合成工艺改进具有较高的研究价值。
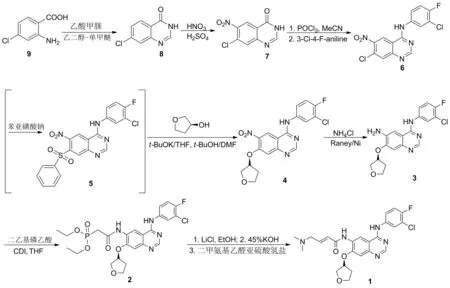
Scheme 1
本文在文献[7,13]方法的基础上,改进了1的合成工艺。以9为原料,经环合、硝化、氯代和胺化后,采用一锅两步法制得关键中间体4-[(3-氯-4-氟苯基)氨基]-6-硝基-7-[(S)-四氢呋喃-3-基氧基]-喹唑啉(4); 4依次经还原、酰胺化、HWE反应合成1,总收率55.7%,含量98%,其结构经1H NMR和LC-MS确证。
1 实验部分
1.1 仪器与试剂
SGW X-4B型熔点仪;ZF-20D型暗箱式紫外分析仪;BrukerAVANCE III 400M型核磁共振仪(DMSO-d6为溶剂,TMS为内标);LC-MS-2020型液质联用仪;LC-3000型液相色谱仪。
本研究显示我院慢阻肺首次确诊重度及以上比例约为50%,慢阻肺平均错失早期诊断时间中位数为3年。慢阻肺患者年龄、错失早期诊断时间与疾病严重程度相关,年龄越大,错失早期诊断时间越长,疾病越严重。
(S)-3-羟基四氢呋喃,常茂生物化学工程股份有限公司,含量99.5%;二甲氨基乙醛亚硫酸氢盐参考文献[7]方法合成;其余所用试剂均为分析纯。
1.2 合成
(1) 7-氯喹唑啉-4-酮(8)的合成
在四口烧瓶中依次加入9 4.5 g(26.2 mmol),乙酸甲脒6.8 g(65.5 mmol)和乙二醇-单醚25 mL,搅拌下回流反应2 h。冷却至室温,倒入冰水中,析出灰白色固体,抽滤,滤饼用水(100 mL)洗涤,真空干燥得灰白色固体8 4.5 g,收率95.7%,含量98%(HPLC,下同),m.p.247~250 ℃;1H NMRδ: 12.33(s, 1H), 8.16(dd,J=8.7 Hz, 6.4 Hz, 1H), 8.12(s, 1H), 7.43(dd,J=10.1 Hz, 2.3 Hz, 1H), 7.37(td,J=8.7 Hz, 2.5 Hz, 1H); APCI-MSm/z: 181{[M+H]+}。
(2) 7-氯-6-硝基喹唑啉-4-酮(7)的合成
搅拌下,在四口烧瓶中加入浓硫酸5 mL,冰盐浴冷却至-5 ℃,缓慢加入发烟硝酸5 mL,分批加入8 3.2 g(17.8 mmol),加毕,反应30 min;于室温反应1 h;升温至100 ℃反应1.5 h。冷却至室温,缓慢倒入冰水中,用饱和碳酸氢钠溶液调至pH 7,析出大量淡黄色固体,抽滤,滤饼用混合溶液[V(EtOH) ∶V(H2O)=1 ∶5](3×50 mL)洗涤,真空干燥得淡黄色固体7 3.0 g,收率77.3%,含量98.3%;1H NMRδ: 12.78(s, 1H), 8.67(s, 1H), 8.30(s, 1H), 8.02(s, 1H); APCI-MSm/z: 224{[M-H]-}。
(3) 4-[(3-氯-4-氟苯基)氨基]-7-氯-6-硝基喹唑啉(6)的合成
在四口烧瓶中加入7 2.3 g(10.2 mmol), DMF 3~4滴和二氯亚砜10 mL,搅拌下回流30 min至固体完全溶解;回流反应2.5 h。蒸除二氯亚砜,残余物用无水二氯甲烷洗涤(3×15 mL)得淡黄色固体。加入3-氯-4-氟苯胺1.8 g(12.2 mmol)的无水1,4-二氧六环(25 mL)溶液,加毕,反应液呈黄色混浊液状,缓慢升温至90 ℃,反应1 h。冷却至室温,倒入冰水中,析出淡橘色固体,用饱和碳酸氢钠水溶液洗至pH 7,抽滤,真空干燥得橘黄色固体6 3.5 g,收率98%,含量96%, m.p.240~243 ℃;1H NMRδ: 10.39(s, 1H), 9.39(s, 1H), 8.74(s, 1H), 8.27~7.96(m, 2H), 7.76(s, 1H), 7.47(t,J=9.0 Hz, 1H); APCI-MSm/z: 352{[M-H]-}。
(4) 4的合成
在单口烧瓶中加入(S)-3-羟基四氢呋喃1.1 g(12.1 mmol)和无水DMF 7 mL,搅拌下冷却至0 ℃,分批加入t-BuOK 3.6 g(32.5 mmol),加毕,反应2 h得反应液A。
在四口烧瓶中依次加入6 3.3 g(9.3 mmol)和苯亚磺酸钠1.7 g(10.23 mmol),搅拌下于90 ℃反应2 h得反应液B。
搅拌下,将A于50 ℃缓慢滴加至B中,反应4 h。冷却至室温,倒入冰水中,析出黄色固体,用1 mol·L-1盐酸调至pH 7,抽滤,滤饼用水多次洗涤,真空干燥得黄色固体4 3.7 g,收率97.8%,含量97.7%, m.p.209~211 ℃;1H NMRδ: 10.19(s, 1H), 9.23(s, 1H), 8.68(s, 1H), 8.17(dd,J=6.8 Hz, 2.5 Hz, 1H), 7.90~7.68(m, 1H), 7.56~7.39(m, 2H), 5.46(s, 1H), 3.98(dd,J=10.6 Hz, 4.2 Hz, 1H), 3.94~3.71(m, 3H), 2.35(td,J=14.3 Hz, 8.4 Hz, 1H), 2.14~1.98(m, 1H); APCI-MSm/z: 403{[M-H]-}。
(5) 4-[(3-氯-4-氟苯基)氨基]-6-氨基-7-[(S)-四氢呋喃-3-基氧基]-喹唑啉(3)的合成
搅拌下,在四口烧瓶中依次加入4 2.0 g(4.9 mmol), FeCl3·6H2O/C 4.0 g和乙醇15 mL,升温至回流,加入水合肼0.63 g(12.3 mmol),反应1 h。旋除溶剂,用少量乙醇重结晶,过滤,滤饼真空干燥得淡黄色粉末3 1.77 g,收率96.0%,含量97.0%, m.p.128~130 ℃;1H NMRδ: 9.42(s, 1H), 8.37(s, 1H), 8.19(dd,J=6.9 Hz, 2.4 Hz, 1H), 7.89~7.71(m, 1H), 7.50~7.27(m, 2H), 7.05(s, 1H), 5.41(s, 2H), 5.24(s, 1H), 4.05~3.85(m, 3H), 3.80(dt,J=12.9 Hz, 6.5 Hz, 1H), 2.32(td,J=14.3 Hz, 7.9 Hz, 1H), 2.19~2.06(m, 1H); APCI-MSm/z: 373{[M-H]-}。
(6) 【2-【{4-[(3-氯-4-氟苯基)-氨基]-7-[(S)-四氢呋喃-3-基)氧基]喹唑啉-6-基}氨基】-2-氧代乙基】磷酸二乙酯(2)的合成
搅拌下,在单口烧瓶中加入二乙基磷乙酸0.94 g(4.8 mmol), DMF 3~4滴和无水二氯甲烷10 mL,冷却至0 ℃,滴加草酰氯0.98 g(7.7 mmol),滴毕,反应2 h。旋蒸除去溶剂得黄色油状液体二乙基磷乙酰氯。
在50 mL单口烧瓶中加入3 1.2 g(3.2 mmol),三乙胺0.65 g(6.4 mmol)和无水二氯甲烷10 mL,于0 ℃滴加二乙基磷乙酰氯0.82 g(3.8 mmol)的无水二氯甲烷(15 mL)溶液,反应1 h。用饱和食盐水(3×100 mL)洗涤,有机相用无水硫酸钠干燥,旋蒸得淡黄色固体2 1.8 g,收率91.0%,含量97.0%; m.p.185~188 ℃;1H NMRδ: 9.91(s, 1H), 9.49(s, 1H), 8.93(s, 1H), 8.52(s, 1H), 8.08(dd,J=6.9 Hz, 2.5 Hz, 1H), 7.82~7.68(m, 1H), 7.43(t,J=9.1 Hz, 1H), 7.25(s, 1H), 5.32(d,J=2.4 Hz, 1H), 4.10(d,J=7.2 Hz, 4H), 4.02(d,J=3.5 Hz, 2H), 3.95(dd,J=15.3 Hz, 7.7 Hz, 1H), 3.80(td,J=8.1 Hz, 4.9 Hz, 1H), 3.42(s, 2H), 2.34(dt,J=14.1 Hz, 7.0 Hz, 1H), 2.18(dd,J=12.5 Hz, 5.8 Hz, 1H), 1.26(t,J=7.0 Hz, 6H); APCI-MSm/z: 551{[M-H]-}。
(7) 1的合成
搅拌下,在单口烧瓶中依次加入2 1.00 g(1.8 mmol),氯化锂77 mg(1.8 mmol)和乙醇10 mL,冷却至-5 ℃,依次缓慢滴加45%KOH溶液1 mL和二甲氨基乙醛亚硫酸氢盐0.49 g(2.9 mmol)的水(5 mL)溶液,滴毕,反应1 h。加入水100 mL,析出白色固体,抽滤,真空干燥得白色固体1 0.8 g,收率90%,含量98.0%; m.p.127~130 ℃;1H NMRδ: 9.83(s, 1H), 9.46(s, 1H), 8.97(s, 1H), 8.53(s, 1H), 8.13(dd,J=6.9 Hz, 2.6 Hz, 1H), 7.80(ddd,J=9.0 Hz, 4.3 Hz, 2.7 Hz, 1H), 7.43(t,J=9.1 Hz, 1H), 7.24(s, 1H), 6.81(dt,J=15.4 Hz, 5.9 Hz, 1H), 6.60(d,J=15.6 Hz, 1H), 5.30(d,J=3.1 Hz, 1H), 4.07~3.88(m, 3H), 3.79(td,J=8.1 Hz, 5.0 Hz, 1H), 3.09(d,J=5.3 Hz, 2H), 2.35(dd,J=13.6 Hz, 6.4 Hz, 1H), 2.19(s, 6H), 2.14(d,J=6.6 Hz, 1H); APCI-MSm/z: 484{[M-H]-}。
2 结果与讨论
2.1 合成
在6的合成中,文献[7]采用三氯氧磷为氯代试剂、乙腈及二氧六环为溶剂,毒性大、后处理不便。我们改用二氯亚砜为氯化剂和溶剂进行氯代反应,经减压蒸馏后处理后与3-氯-4-氟苯胺反应,使该步反应收率提高至98%(89.6%[7]),后处理也较方便。
在4的合成中,文献[7]先用6与苯亚磺酸钠反应制得4-[(3-氯-4-氟苯基)氨基]-6-硝基-7-苯磺酰基喹唑啉(5); 5在催化剂作用下,与(S)-3-羟基-四氢呋喃在多种溶剂中反应合成4。该方法存在步骤多、溶剂复杂,后处理繁琐等缺点。我们改用一锅法反应,4与苯亚磺酸钠反应后不经处理,在叔丁醇钾作用下,直接与(S)-3-羟基-四氢呋喃反应,操作简便,收率提高至97.8%(77%[7])。
在2的合成中,文献[7]选择在CDI作用下,3与二乙基磷乙酸反应合成2,收率较低,反应时间较长。我们改用草酰氯先与二乙基磷乙酸反应先制得酰氯,再与3反应合成2,收率提高至91.0%(69.8%[7])。
2.2 4的合成条件优化
我们发现,用“一锅法”合成4时,第二步反应对反应影响较大。因此,研究了温度、催化剂种类及其用量对反应的影响,结果见表1。
表1 醚化反应条件优化
Table 1 Optimization of reaction conditions for etherification
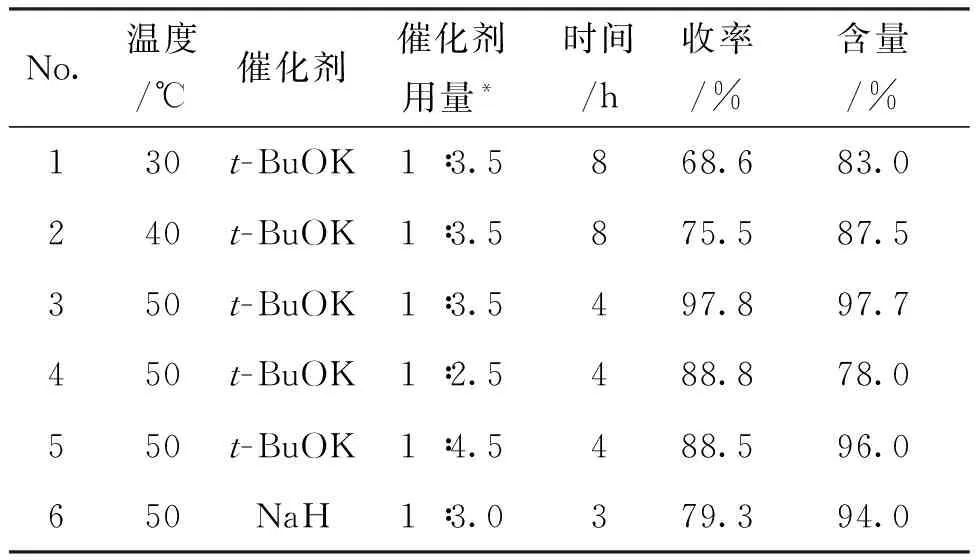
No.温度/℃催化剂催化剂用量*时间/h收率/%含量/%130t-BuOK1∶3.5868.683.0240t-BuOK1∶3.5875.587.5350t-BuOK1∶3.5497.897.7450t-BuOK1∶2.5488.878.0550t-BuOK1∶4.5488.596.0650NaH1∶3.0379.394.0
*n(6) ∶n(催化剂)。
由表1可以看出,No.1~No.3为反应温度对反应的影响,反应温度为50 ℃时,反应效果最好(No.3)。 No.3和No.6为催化剂种类对反应的影响,选用t-BuOK作催化剂,收率和含量最高,以NaH为催化剂,虽然可以缩短反应时间,但收率较低(No.6)。 No.3~No.5为催化剂用量对反应的影响,当催化剂用量为1 ∶3.5时,反应效果最好。
综合考虑,醚化反应的最优条件为:t-BuOK为催化剂(1 ∶3.5),于50 ℃反应4 h, 4收率97.8%,含量97.7%。
[1] 王允芬,宋勇. Afatinib(阿法替尼)治疗非小细胞肺癌的研究进展[J].中华肺部疾病杂志,2012,05(04):55-60.
[2] US FDA. FDA approves new treatment for a type oflatestage lung cancer[EB/OL].[2014-05-12]. http://www.fda.gov/NewsEvents/Newstroom/PressAnnouncements/ucm360499.html
[3] 陈庆财,赵俊,赵小伟,等. 一种阿法替尼化合物的制备方法:CN 103755688A[P].2014.
[4] Himmelsbach F, Langkopf E, Blech S,etal. Quinazolinederivatives,medicaments containing said compounds,their utilization and method for the production thereof:WO 0250043A1[P].2002.
[5] Cha M Y, Lee K O, Kim J W,etal. Discovery of a novel Her-1/Her-2 dual tyrosine kinase inhibitor for the treatment of Her-1 selective inhibitor-resistant non-small cell lung cancer[J].Journal of Medicinal Chemistry,2009,52(21):6880-6888.
[6] Rall W, Soyka R, Sieger R,etal. Method for the production of aminocrotonylcompounds:WO 2005037824A2 [P].2005.
[7] Schroeder J, Dziewas G, Fachinger T,etal. Process for preparing aminocrotonylamino-substituted quinazolinederivatives:WO 2007085638A1[P].2007.
[8] Schroeder J, Dziewas G, Fachinger T,etal. Process for preparing aminocrotonylamino-substituted quinazolinederivatives:US 8067593[P].2011.
[9] Albrecht W, Fischer D, Gidwani R M,etal. Novel salts and polymorphic forms of afatinib:WO 2012121764A1[P].2012.
[10] Xu X. Methodfor preparing afatinib and intermediate thereof:WO 2014180271[P].2014.
[11] Xu X. Afatinib and preparation method of intermediate thereof:WO 2014183560[P].2014.
[12] Tuskar M, Ratkaj M, Zegarac M. Crystalline froms of Afatinibdimaleate:WO 2015103456[P].2015.
[13] 李晴晴,张庆文. 阿法替尼的合成路线图解[J].中国医药工业杂志,2015,46(04):422-424.
Process Improvement on the Synthesis of Afatinib
LI Wen-qian1, SONG Guo-qiang1, WAN Yi-dong2,GAO You-jun2, FENG Xiao-qing2,3*
(1. School of Pharmaceutical Engineering and Life Sciences, Changzhou University, Changzhou 213164, China;2. Changmao Biochemical Engineering Company Ltd., Changzhou 213034, China;3. College of Chemical Engineering, Nanjing Tech University, Nanjing 210009, China)
The key intermediate, 4-[(3-Chloro-4-fluorophenyl)amino]-6-nitro-7-[(S)-tetrahydrofuran-3-yloxy]quinazolin(4), was prepared from 2-amino-4-chlorobenzoic acid by cyclization, nitrification, chlorine generation and amination, then a “one-pot” process. Afatinib with total yield of 55.7% and purity of 98% was synthesized by reduction, amidation and HWE from 4. The structure was confirmed by1H NMR and LC-MS.
Afatinib; drug synthesis; one-pot method; process improvement
2016-10-09;
2016-12-07
江苏省博士后科研资助计划(1501128B)
李文倩(1991-),女,汉族,江苏常州人,硕士研究生,主要从事药物合成工艺的研究。 E-mail: 374191893@qq.com
冯筱晴,讲师, E-mail: fxqfw@163.com
R914.5; O623.7
A
10.15952/j.cnki.cjsc.1005-1511.2017.02.16253
猜你喜欢
化工设计(2022年4期)2023-01-02
贵州科学(2022年4期)2022-09-05
天然产物研究与开发(2018年8期)2018-09-10
化工管理(2017年35期)2018-01-10
益寿宝典(2017年1期)2017-09-03
中小学实验与装备(2016年1期)2016-04-19
中国资源综合利用(2016年7期)2016-02-03
浙江理工大学学报(自然科学版)(2015年7期)2015-03-01
红领巾·探索(2014年8期)2014-10-10
火炸药学报(2014年5期)2014-03-20
