
新型常春藤皂苷元衍生物的合成及其体外抗HBV活性
2017-02-07 08:59洪开文贾宪生杨贤江董登祥
合成化学 2017年1期
洪开文, 贾宪生, 杨贤江, 董登祥*
(1. 安顺职业技术学院 应用医药系,贵州 安顺 561000; 2. 贵阳中医学院 药学院,贵州 贵阳 550001)
·研究论文·
新型常春藤皂苷元衍生物的合成及其体外抗HBV活性
洪开文1, 贾宪生2, 杨贤江2, 董登祥2*
(1. 安顺职业技术学院 应用医药系,贵州 安顺 561000; 2. 贵阳中医学院 药学院,贵州 贵阳 550001)
以常春藤皂苷元为原料,对其3,23-羟基和28-羧基进行乙酰化、醚化、苯甲酰化及酯化等反应合成了8个常春藤皂苷元衍生物(2~9),其中6个未见文献报道,其结构经1H NMR,13C NMR, IR和MS表征。采用MTT法测定了2~9的体外抗HBV活性。结果表明:3~9随浓度增大,对细胞活力的抑制率明显增加,特别是7和9抑制率最明显,用药浓度为1×10-6mol·L-1时,抑制率均为9.46±0.47%。对2~9抑制Hep G2.2.15细胞表达表面抗原(HBs Ag)进行了测试。结果表明:2~9对HBs Ag的分泌表达有一定的抑制作用。
常春藤皂苷元; 衍生物; 合成; 抗HBV活性
常春藤皂苷元(Chart 1)及其糖苷在自然界分布相对较广,但是其在植物中的含量一般较低,而含量高的植物则相对较少。研究表明,常春藤皂苷元具有抗炎、抗抑郁及抗肿瘤等药理作用[1-5]。孙璐[6]报道以异珠五加甲苷为原料合成了常春藤皂苷元衍生物,该类衍生物在体外对肝癌细胞有较好的抑制活性。王国华[7]设计并合成了常春藤皂苷元酰胺衍生物,首先用N,N′-二环己基碳二亚胺(DCC)、N-羟基琥珀酰亚胺(NHS)形成活化酯,再加入3-二甲氨基丙胺进行反应,合成了目标化合物N-(3-二甲氨基丙基)-常春藤皂苷元-17-甲酰胺(HGA),研究表明结构改造后的化合物HGA的抗抑郁生物活性显著提高。Martin等[8]报道合成了六个商业二糖(D-乳糖、D-纤维二糖、D-麦芽糖、D-蜜二糖、D-龙胆二糖、D-异麦芽糖)的常春藤皂苷衍生物。毛玝佳等[9]报道常春藤皂苷元在Py/Ac2O, MeI/TBAF等条件下,合成了常春藤皂苷元二乙酰化衍生物A、常春藤皂苷元羧基甲酯衍生物B和常春藤皂苷元乙酰化羧甲酯衍生物C。吴耀民等[10]报道将常春藤皂苷元的95%乙醇溶液分别加至NaOH和KOH的70%乙醇溶液中,用95%乙醇重结晶制得常春藤皂苷元钠和常春藤皂苷元钾。
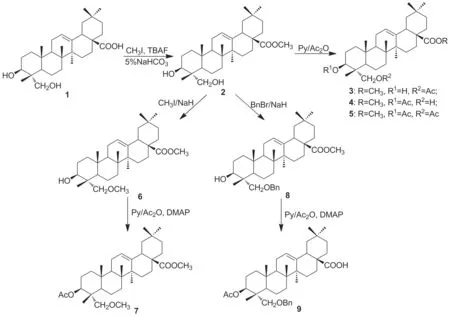
Scheme 1
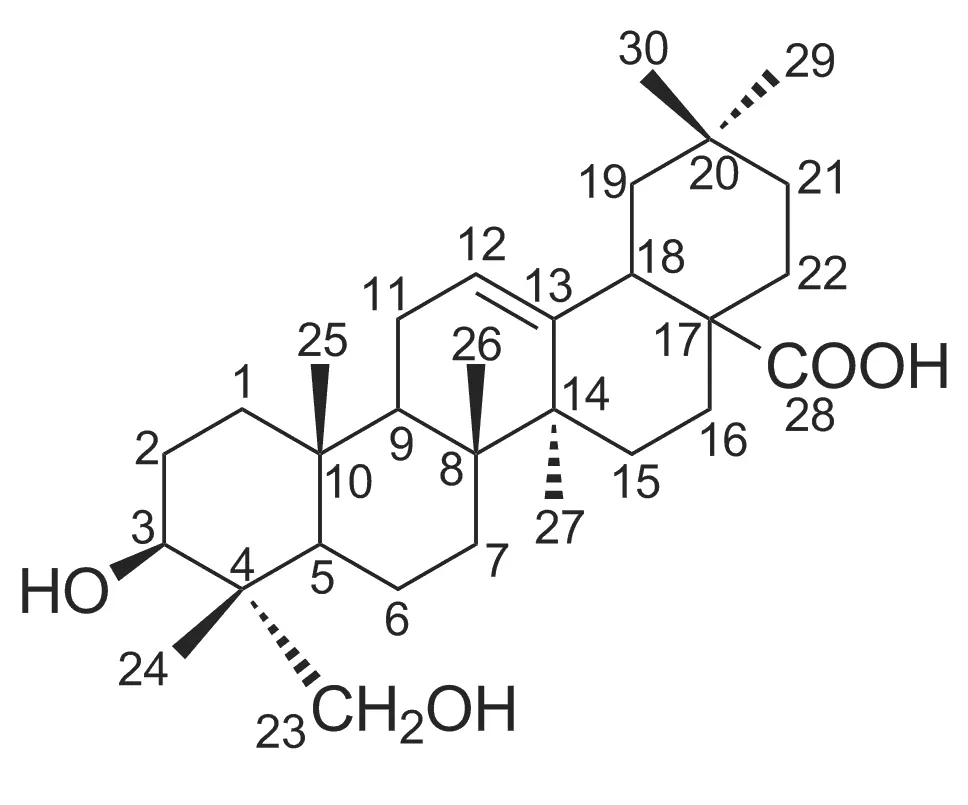
Chart 1
此外,贵州山银花黄褐毛忍冬为双子叶植物药忍冬科植物黄褐毛忍冬(LonicerafulvotnetosaHsu et S. C. Cheng. Ms)的花蕾[11],具有清热解毒和疏散风热的功效,用于治疗臃肿疔疮、喉痹、丹毒、热毒血痢、风热感冒及温热发病。在贵州分布广,资源极其丰富,其所含化学成分有常春藤皂苷及苷元和绿原酸等[12],为本课题研究提供大量原料。
本文设计了关于常春藤皂苷元的结构改造途径。以常春藤皂苷元(1)为原料,经酯化、乙酰化、苯甲酰化等反应,合成了8个常春藤皂苷元衍生物(2~9, Scheme 1),其中化合物3, 4, 6~9未见文献报道,其结构经1H NMR,13C NMR, IR和MS表征。采用MTT法测定了化合物2~9的体外抗HBV活性,进行化合物抑制Hep G2. 2. 15细胞表达表面抗原(HBs Ag)的测试。为进一步研究常春藤皂苷元衍生物的体内活性打下基础。
1 实验部分
1.1 仪器与试剂
XCL-1型显微熔点仪;1801型紫外-可见分光光度计;INOVA-400型与INOVA-500型核磁共振仪(CDCl3为溶剂,TMS为内标);WGH-30型双光束红外分光光度计;FINNIGANLACQ-DECA型质谱仪。
常春藤皂苷元由贵阳中医学院贾宪生教授提供;常春藤皂苷元对照品,纯度≥99%,西安昊轩生物科技有限公司;溴化苄,化学纯,国药集团化学试剂有限公司;碘甲烷,分析纯,成都市科龙化工试剂有限公司;其余所用试剂均为分析纯。
体外抗HBV活性实验委托贵州省中国科学院天然产物化学重点实验室药理与活性筛选中心完成。
1.2 合成
(1) 1的提取分离
将0.5 kg粉碎黄褐毛忍冬干燥花蕾用75%乙醇回流提取3次,合并提取液,减压回收乙醇得残留物150 mL,加5%盐酸,置水浴中水解3 h(TLC监测至无皂苷斑点)。冷却后过滤,沉淀用水洗至中性,干燥得总皂苷元14.6 g。
取总皂苷元2.0 g,用少量无水乙醇溶解,加入适量硅胶搅拌,水浴上挥干,残渣研细,经硅胶H柱层析[洗脱剂:氯仿/甲醇(V/V=9/1)]分离纯化,合并3~7份洗脱液,回收溶剂,残留物用甲醇结晶得白色短柱状晶体1,收率27.5%,Rf=0.42[展开剂:A=Cy/EtOAc(V/V=1/2)],与对照品一致, m.p.132~134 ℃;1H NMR(400 MHz)δ: 5.26(m, 1H, 12-H), 3.72(d,J=11.0 Hz, 2H, 23-H), 3.68(m, 1H, 3-H), 3.28(m, 1H, 18-H), 1.25(s, 3H, 27-H), 1.05(s, 3H, 26-H), 1.00(s, 3H, 30-H), 0.88(s, 3H, 24-H), 0.79(s, 3H, 29-H), 0.74(s, 3H, 25-H); IRν: 3 451(OH), 2 942(CH), 1 698(C=O), 1 464, 1 387, 1 303, 1 268(C—O) cm-1。以上数据与常春藤皂苷元的表征数据一致[13],故将1鉴定为常春藤皂苷元。
(2) 2的合成
在反应瓶中依次加入1 500 mg(1.05 mmol),碘甲烷4 mL(6.4 mmol),氟化四丁基铵(TBAF)167 mg(0.64 mmol), 5%NaHCO3溶液40 mL和DCM 20 mL, N2保护,搅拌下于室温反应24 h(TLC监测,展开剂:A=1/1,Rf=0.52)。用DCM萃取,萃取液用无水MgSO4干燥,减压浓缩得粗产物,经硅胶柱层析(洗脱剂:A=2/1)纯化得白色固体粉末2 490 mg,收率95.1%, m.p.144~145 ℃;1H NMR(400 MHz)δ: 5.28(m, 1H, 12-H), 3.73(d,J=10.4 Hz, 2H, 23-H), 3.64(m, 1H, 3-H), 3.62(s, 3H, OCH3), 2.85(dd,J=13.2 Hz, 3.6 Hz, 1H, 18-H), 2.56(br s, 1H, OH), 2.23(br s, 1H, OH), 1.96(m, 2H, 11-H), 1.78(m, 2H, 2-H), 1.67(m, 2H, 22-H), 1.47(m, 2H, 16-H), 1.42(m, 2H, 5, 9-H), 1.38(m, 6H, 1,6,7-H), 1.34(m, 4H, 19,21-H), 1.23(m, 2H, 15-H), 1.12(s, 3H, 27-H), 0.98(s, 3H, 26-H), 0.95(s, 3H, 30-H), 0.89(s, 3H, 24-H), 0.79(s, 3H, 29-H), 0.73(s, 3H, 25-H)。表征数据与常春藤皂苷-28-羧甲酯的数据[14]一致。
(3) 3~5的合成
在反应瓶中依次加入2 100 mg(0.21 mmol),吡啶(Py)2 mL(0.02 mol)和醋酸酐1 mL(10.61 mmol), N2保护,搅拌下于室温反应1.5 h(TLC检测,展开剂:A=3/1)。加入1 mol·L-1盐酸调至pH 7,用二氯甲烷萃取,萃取液用无水MgSO4干燥,减压浓缩得粗产物,经硅胶柱层析(洗脱剂:A=5 ∶1)纯化得3~5。
3: 白色固体粉末35 mg,收率32.4%, m.p.153~155 ℃;1H NMR(400 MHz)δ: 5.28(m, 1H, 12-H), 3.82(d,J=11.2 Hz, 2H, 23-H), 3.62(s, 3H, OCH3), 3.59(m, 1H, 3-H), 2.85(dd,J=14.0 Hz, 4.4 Hz, 1H, 18-H), 2.14(s, 3H, COCH3), 2.06(m, 2H, 11-H), 1.71(m, 2H, 2-H), 1.65(m, 2H, 22-H), 1.45(m, 2H, 16-H), 1.41(m, 2H, 5,9-H), 1.39(m, 6H, 1,6,7-H), 1.35(m, 4H, 19,21-H), 1.24(m, 2H, 15-H), 1.16(s, 3H, 27-H), 0.98(s, 3H, 26-H), 0.94(s, 3H, 30-H), 0.88(s, 3H, 24-H), 0.77(s, 3H, 29-H), 0.72(s, 3H, 25-H); IRν: 3 458(OH), 2 944(CH), 2 879(CH3), 1 769(C=O), 1 720 (C=O), l 654(C=C), 1 236(C—O) cm-1; MS(ESI)m/z: 552.3{[M+Na]+}; HR-MSm/z: Calcd for C33H52O5{[M-Bn]-}437.862 6, found 437.861 9。
4: 白色固体粉末20 mg,收率18.5%, m.p.153~154 ℃;1H NMR (400 MHz)δ: 5.28(m, 1H, 12-H), 4.88(m, 1H, 3-H), 3.63(d,J=11.3 Hz, 2H, 23-H), 3.62(s, 3H, OCH3), 2.85(dd,J=15.2 Hz, 5.6 Hz, 1H, 18-H), 2.18(s, 3H, COCH3), 2.07(m, 2H, 11-H), 1.73(m, 2H, 2-H), 1.69(m, 2H, 22-H), 1.46(m, 2H, 16-H), 1.42(m, 2H, 5, 9-H), 1.37(m, 6H, 1,6,7-H), 1.33(m, 4H, 19,21-H), 1.21(m, 2H, 15-H), 1.16(s, 3H, 27-H), 0.96(s, 3H, 26-H), 0.92(s, 3H, 30-H), 0.88(s, 3H, 24-H), 0.72(s, 3H, 29-H), 0.67(s, 3H, 25-H); IRν: 3 448(OH), 2 945(CH), 2 894(—CH3), 1 760(C=O), 1 726(C=O), l 656(C=C), 1 239(C—O) cm-1; MS(ESI)m/z: 552.2{[M+Na]+}; HR-MSm/z: Calcd for C33H52O5{[M-Bn]-}437.762 5, found 437.761 8。
5: 白色固体56 mg,收率47.8%, m.p.163~164 ℃;1H NMR (400 MHz)δ: 5.28(m, 1H, 12-H), 4.78(m, 1H, 3-H), 3.69(d,J=11.6 Hz, 2H, 23-H), 3.62(s, 3H, OCH3), 2.86(dd,J=14.0 Hz, 4.4 Hz, 1H, 18-H), 2.17(s, 3H, COCH3), 2.09(s, 3H, COCH3), 1.97(m, 2H, 11-H), 1.72(m, 2H, 2-H), 1.67(m, 2H, 22-H), 1.45(m, 2H, 16-H), 1.43(m, 2H, 5,9-H), 1.36(m, 6H, 1,6,7-H), 1.34(m, 4H, 19,21-H), 1.21(m, 2H, 15-H), 1.16(s, 3H, 27-H), 0.96(s, 3H, 26-H), 0.92(s, 3H, 30-H), 0.88(s, 3H, 24-H), 0.80(s, 3H, 29-H), 0.72(s, 3H, 25-H);13C NMR (101 MHz)δ: 178.2(C28), 170.7, 171.0(2C, COCH3), 143.8(C13), 122.1(C12), 74.5(C3), 65.4(C23), 51.5(OCH3), 47.8(C9), 47.6(C5), 46.6(C17), 45.7(C19), 41.5(C4), 41.2(C14), 40.4(C18), 39.2(C8), 37.6(C1), 36.7(C10), 33.8(C21), 33.0(C29), 32.3(C22), 32.2(C7), 30.6(C20), 27.6(C15), 25.7(C27), 23.6(C2), 23.3(C11), 22.9(C30), 21.2(C1), 20.9(2C, COCH3), 17.9(C6), 16.8(C26), 15.7(C25), 13.0(C24)。以上数据与常春藤皂苷元-3,23-二-O-乙酰基-28-羧甲酯的数据[14]一致,故将5鉴定为常春藤皂苷元-3,23-二-O-乙酰基-28-羧甲酯。
(4) 6的合成
在反应瓶中依次加入2 80 mg(0.16 mmol)和4 Å分子筛30 mg,抽真空,N2保护,加入无水DCM 5 mL,冰浴(0 ℃)冷却下搅拌平衡10 min;加入CH3I 1 mL(16.06 mmol)和60%NaH 59 mg(4.10 mmol),于0 ℃反应8 h(TLC监测,展开剂:A=2/1,Rf=0.61)。加入适量甲醇中和过量NaH,用二氯甲烷萃取,萃取液用无水MgSO4干燥,减压浓缩得粗产物,经硅胶柱层析(洗脱剂:A=5/1)纯化得黄白色固体粉末6 61 mg,收率74.82%, m.p.146~147 ℃;1H NMR(400 MHz)δ: 5.28(m, 1H, 12-H), 3.57(d,J=10.4 Hz, 1H, 23-H), 3.62(s, 3H, OCH3), 3.34(s, 3H, OCH3), 3.17(m, 1H, 3-H), 2.85(dd,J=14.0 Hz, 4.4 Hz, 1H, 18-H), 1.97(m, 2H, 11-H), 1.72(m, 2H, 2-H), 1.67(m, 2H, 22-H), 1.45(m, 2H, 16-H), 1.43(m, 2H, 5,9-H), 1.36(m, 6H, 1,6,7-H), 1.34(m, 4H, 19,21-H), 1.21(m, 2H, 15-H), 1.16(s, 3H, 27-H), 0.92(s, 3H, 26-H), 0.89(s, 3H, 30-H), 0.88(s, 3H, 24-H), 0.84(s, 3H, 29-H), 0.71(s, 3H, 25-H);13C NMR(101 MHz)δ: 178.2(C28), 143.6(C13), 122.2(C12), 84.0(C23), 70.0(C3), 59.4(OCH3), 51.5(OCH3), 49.2(C9), 47.6(C5), 46.6(C17), 45.8(C19), 41.8(C4), 41.6(C14), 41.3(C18), 39.2(C8), 37.9(C1), 36.8(C10), 33.8(C21), 33.0(C29), 32.4(C22), 32.3(C7), 30.6(C20), 27.6(C15), 25.8(C27), 23.6(C2), 23.3(C11), 23.0(C30), 21.2(C16), 18.1(C6), 16.8(C26), 15.6(C25), 12.0(C24); IRν: 3 426(OH), 2 945(CH), 2 845(CH3), 2 823(OCH3), 1 721(C=O), l 653(C=C), 1 232(C—O) cm-1; MS(ESI)m/z: 523.2{[M+Na]+}, HR-MSm/z: Calcd for C32H52O4{[M-Bn]-}408.762 4, found 408.761 8。
(5) 7的合成
在反应瓶中依次加入6 51 mg(0.10 mmol), Py 2 mL(0.02 mol), Ac2O 1 mL(10.61 mmol)和对二甲氨基吡啶(DMAP)10 mg(0.02 mmol), N2保护,搅拌下于室温反应8 h(TLC监测,展开剂:A=3/1,Rf=0.72)。加入1 mol·L-1盐酸调至中性,用二氯甲烷萃取,萃取液用无水MgSO4干燥,减压浓缩得粗产物,经硅胶柱层析(洗脱剂:A=8/1)纯化得淡黄白色固体7 54 mg,收率98.2%, m.p.151~152 ℃;1H NMR(400 MHz)δ: 5.28(m, 1H, 12-H), 3.69(d,J=10.2 Hz, 2H, 23-H), 3.62(s, 3H, OCH3), 3.25(s, 3H, OCH3), 3.18(m, 1H, 3-H), 2.85(dd,J=13.7 Hz, 4.4 Hz, 1H, 18-H), 2.05(s, 3H, COCH3), 1.96(m, 2H, 11-H), 1.71(m, 2H, 2-H), 1.66(m, 2H, 22-H), 1.46(m, 2H, 16-H), 1.44(m, 2H, 5,9-H), 1.37(m, 6H, 1,6,7-H), 1.34(m, 4H, 19,21-H), 1.21(m, 2H, 15-H), 1.12(s, 3H, 27-H), 0.92(s, 3H, 26-H), 0.89(s, 3H, 30-H), 0.88(s, 3H, 24-H), 0.84(s, 3H, 29-H), 0.71(s, 3H, 25-H);13C NMR(101 MHz)δ: 178.2(C28), 170.5(COCH3), 143.7(C13), 122.2(C12), 81.3(C3), 75.0(C23), 59.0(OCH3), 51.4(OCH3), 47.9(C9), 47.3(C5), 46.6(C17), 45.7(C19), 41.6(C4), 41.4(C14), 41.2(C18), 39.1(C8), 37.6(C1), 36.8(C10), 33.7(C21), 33.0(C29), 32.3(C22), 32.1(C7), 30.6(C20), 27.6(C15), 25.7(C27), 23.5(C2), 23.3(C11), 23.0 (C30), 21.2 (C16), 20.9(COCH3), 17.7(C6), 16.7(C26), 15.6 (C25), 13.3(C24); IRν: 2 924(CH), 2 893(CH3), 2 826(OCH3), 1 768(C=O), 1 721(C=O), l 645(C=C), 1 238(C—O) cm-1; MS(ESI)m/z: 565.3{[M+Na]+}; HR-MSm/z: Calcd for C34H54O5{[M-Bn]-}450.862 3, found 450.861 7。
(6) 8的合成
在反应瓶中依次加入2 100 mg(0.21 mmol)和4 Å分子筛30 mg,抽真空,N2保护,加入无水DCM 5 mL,于0 ℃冰浴冷却下搅拌平衡10 min;加入溴化苄1 mL(8.42 mmol)和60%NaH 69 mg(4.79 mmol),升至室温,反应8 h(TLC监测,展开剂:A=2/1,Rf=0.64)。加入适量甲醇中和过量NaH,用DCM萃取,萃取液用无水MgSO4干燥,减压浓缩得粗产物,经硅胶柱层析(洗脱剂:A=5/1)纯化得淡黄白色固体8 102 mg,收率86.4%, m.p.221~223 ℃;1H NMR(400 MHz)δ: 7.28~7.35(m, 5H, PhH), 5.28(m, 1H, 12-H), 4.65(d,J=12.0 Hz, 1H, PhCH2), 4.37(d,J=12.0 Hz, 1H, PhCH2), 3.66(d,J=11.2 Hz, 2H, 23-H), 3.62(s, 3H, OCH3), 3.22(m, 1H, 3-H), 2.86(d,J=13.6 Hz, 1H, 18-H), 2.05(m, 2H, 11-H), 1.72(m, 2H, 2-H), 1.67(m, 2H, 22-H), 1.44(m, 2H, 16-H), 1.43(m, 2H, 5,9-H), 1.37(m, 6H, 1,6,7-H), 1.34(m, 4H, 19,21-H), 1.24(m, 2H, 15-H), 1.14(s, 3H, 27-H), 1.12(s, 3H, 26-H), 0.89(s, 3H, 30-H), 0.88(s, 3H, 24-H), 0.84(s, 3H, 29-H), 0.72(s, 3H, 25-H);13C NMR(101 MHz)δ: 178.2(C28), 143.5(C13), 139.3(C1′, Ph), 128.3(C3′,5′, Ph), 127.6(C2′,6′, Ph), 127.3(C4′, Ph), 122.2(C12), 81.5(PhCH2), 70.6(C3), 68.2(C23), 51.5(OCH3), 48.4(C9), 47.5(C5), 46.6(C17), 45.8(C19), 42.1(C4), 41.3(C14), 41.1(C18), 39.2(C8), 37.8(C1), 36.8(C10), 33.8(C21), 33.2(C29), 32.6 (C22), 32.3(C7), 30.6(C20), 27.6(C15), 25.9(C27), 23.6(C2), 23.5(C11), 23.0 (C30), 21.9(C16), 18.0(C6), 16.7(C26), 15.8(C25), 12.4(C24); IRν: 3 438(OH), 3 115(Ph), 2 944(CH), 1 839(CH3), 1 724(C=O), l 646(C=C), 1 578(Ph), 1 236(C—O), 1 158, 762(Ar—H) cm-1; MS(ESI)m/z: 599.5{[M+Na]+}; HR-MSm/z: Calcd for C38H56O4{[M-Bn]-}485.062 8, found 485.062 1。
(7) 9的合成
在反应瓶中依次加入8 52 mg(0.09 mmol), Py 2 mL(0.02 mol), Ac2O 1 mL(10.61 mmol)和DMAP 12 mg(0.02 mmol), N2保护,搅拌下于室温反应8 h(TLC监测,展开剂:A=3/1,Rf=0.73)。加入1 mol·L-1盐酸调至中性,用二氯甲烷萃取,萃取液用无水MgSO4干燥,减压浓缩得粗产物,经硅胶柱层析(洗脱剂:A=8/1)纯化得淡黄白色固体9 54 mg,收率96.9%, m.p.231~233 ℃;1H NMR (400 MHz)δ: 7.26~7.31(m, 5H, Ph), 5.28(m, 1H, 12-H), 4.44(2H, d,J=12.0 Hz, PhCH2), 3.68(d,J=11.5 Hz, 2H, 23-H), 3.62(3H, s, OCH3), 3.25(1H, m, 3-H), 2.86(dd,J=14.1 Hz, 4.0 Hz, 1H, 18-H), 1.92(s, 3H, COCH3), 2.04(m, 2H, 11-H), 1.73(m, 2H, 2-H), 1.66(m, 2H, 22-H), 1.43(m, 2H, 16-H), 1.43(m, 2H, 5,9-H), 1.38(m, 6H, 1,6,7-H), 1.34(m, 4H, 19,21-H), 1.24(m, 2H, 15-H), 1.14(s, 3H, 27-H), 1.05(s, 3H, 26-H), 0.93(s, 3H, 30-H), 0.95(s, 3H, 24-H), 0.89(s, 3H, 29-H), 0.72(s, 3H, 25-H);13C NMR(101 MHz)δ: 178.2(C28), 170.5(COCH3), 143.7(C13), 138.7(C1′, Ph), 128.1(C3′,5′, Ph), 127.5(C2′,6′, Ph), 127.2(C4′, Ph), 122.2(C12), 74.9(C3), 72.7(PhCH2), 71.7(C23), 51.4(OCH3), 47.4(C9), 47.1(C5), 46.6(C17), 45.7(C19), 41.6(C4), 41.2(C14), 41.1(C18), 39.2(C8), 37.6(C1), 36.6(C10), 33.8(C21), 33.0(C29), 32.3(C22), 32.1(C7), 30.6(C20), 27.6(C15), 25.8(C27), 23.6(C2), 23.3(C11), 23.0(C30), 22.9(C16), 21.2(COCH3), 17.7 (C6), 16.7(C26), 15.7 (C25), 13.4(C24); IRν: 3 105(Ph—H), 2 924(CH), 1 849(CH3), 1718(C=O), l 646(C=C), 1 568(Ph), 1 236(C—O), 772(Ar—H) cm-1; MS(ESI)m/z: 627.3{[M+Na]+}; HR-MSm/z: Calcd for C39H56O5{[M-Bn]-}512.862 8, found 512.862 2。
1.3 溶解度测定
取带有恒温夹套的小容量瓶(10 mL)18个,其中1~9号加入适量的蒸馏水,10~18号加入适量乙醚。于25 ℃分别加入适量化合物1~9至饱和,启动电磁搅拌器,恒温下持续搅拌,由于容量瓶容积很小,且电磁搅拌的强度大,因此容量瓶各处可视为无浓度梯度。实验过程中体系始终处于两相共存的状态,1 h后认为体系液相中各个化合物的浓度即为其在该温度下的溶解度[15-16]。为确保溶解平衡的建立,停止搅拌后,静置1 h,使各化合物未溶解部分沉淀到小容量瓶底部,用注射器抽取少量上清液,分别以蒸馏水和乙醚做空白对照,通过紫外-可见分光光度计测定各化合物饱和溶液的吸光度,同时测定化合物标准溶液的吸光度,计算得化合物的溶解度(表 1)。
1.4 体外抗HBV活性测定
首先用MTT法评价化合物在预设浓度下对细胞生长的抑制程度,确定合适的2~9浓度,再进行化合物抑制Hep G2.2.15 细胞表达表面抗原(HBs Ag)的测试。通过计算化合物对HBs Ag表达的抑制率来评价化合物活性。按下式分别计算化合物对细胞活力和对表面抗原的抑制率。
抑制率=[(OD对照-OD样品)/OD对照]×100%
2 结果与讨论
2.1 合成
在3~5的合成中,主要是控制反应时间,研究发现当时间超过3 h后产物只有5;如果时间在30 min以下原料反应不完全,TLC检测只有两个产物点;经过多次试验探讨最终确定反应时间为1.5 h。出现这样的结果,可能是因为常春藤皂苷元的3-位羟基和23-位羟基的活性或空间位阻不同,也为糖基化反应提供参考依据,同时注意吡啶和醋酸酐的体积比例为2 ∶1,因为该条件下合成化合物产率较高。
在7的合成中,由于NaH加入后反应剧烈,而且碘甲烷易挥发,所以该反应需严格控制反应温度在0 ℃冰浴中且避光,产率相对较高。对于反应过量的NaH用适量甲醇中和,研究结果发现只有23-位羟基反应,而3-位羟基没参与反应,以乙酰化产物有区别,分析可能是3-羟基空间位阻较大,具体机理有待进一步研究。
据文献[17]报道先将芳香酸的羧基与甲醇在对甲基苯磺酸作用下形成甲酯,用苄基保护羟基,然后在氢氧化钾的甲醇溶液中水解甲酯得苄基保护酚羟基的芳香酸,但该法酯化反应需较长时间,且产率仅为75%。本文直接将常春藤皂苷元28-位羧基与碘化钾、氟化四丁基铵反应得2,再加入溴化苄、NaH继续反应得8。方法不仅可缩短时间,还使产率提高至86.4%。结果显示只有23-位羟基反应,而3-位羟基未反应,可能是空间位阻的原因。
2.2 溶解度
化合物1~9的溶解度见表1。由表1可以看出,1的水溶性和脂溶性都不好,而其衍生物的溶解性有所改变。在25 ℃时,通过水和乙醚中的溶解结果分析,可以看出5, 7, 9在水中极难溶解,而极易溶于乙醚中,而2~4, 6和8溶解于乙醚,不溶于水。由此可见,合成的常春藤皂苷元衍生物脂溶性均增大,其脂溶性的变化与活性的关系有待进一步研究。
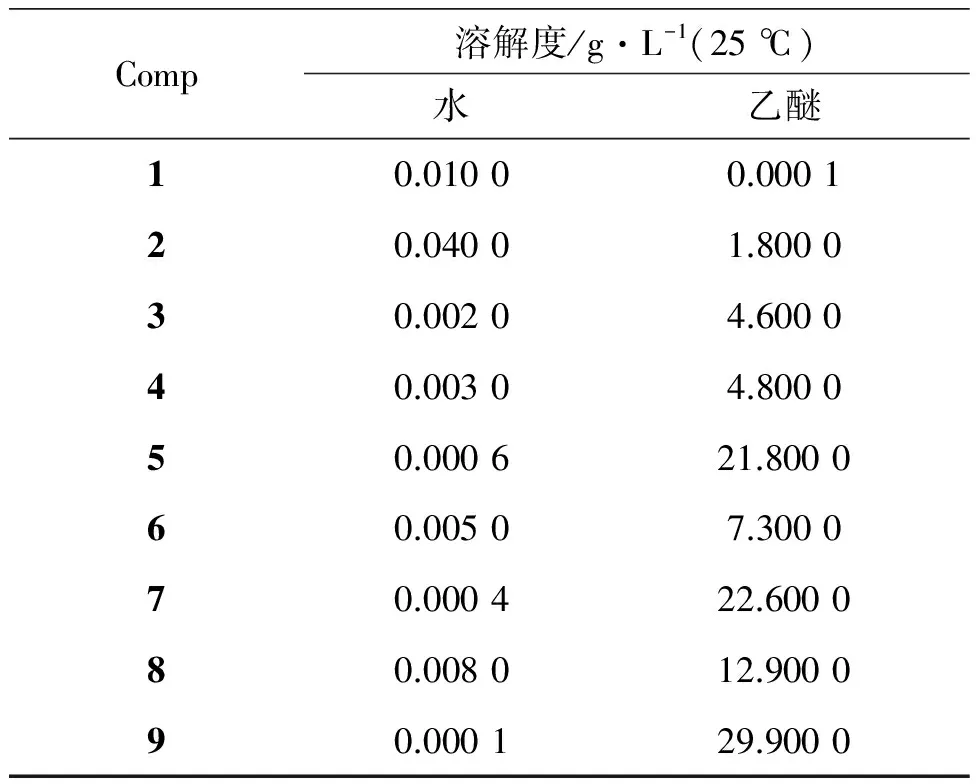
表1 1~9的溶解度
2.3 抗HBV活性
考察了化合物2~9的体外抗HBV活性,结果见表2。由表2可以看出,当浓度为10-6~10-4mol·L-1时,2对细胞活力的抑制率为0,但浓度为10-4mol·L-1时对表面抗原表现有一定抑制率。当然,随着化合物分子量及脂溶性的增加,3~9随浓度的增大对细胞活力的抑制率明显增加,特别是7和9最明显,用药浓度为1×10-6mol·L-1,抑制率均为9.46±0.47%。
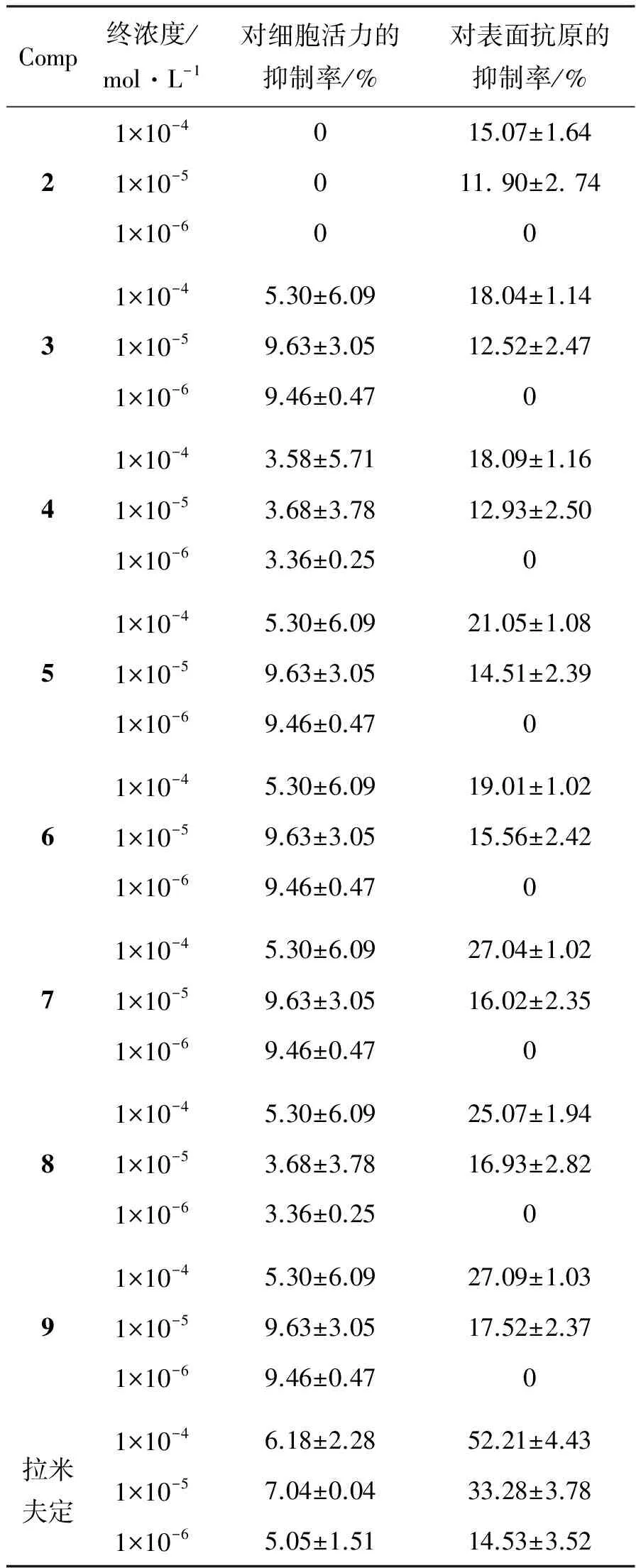
表2 2~9的筛选结果
通过表面抗原的抑制率测试结果分析可知,在一定浓度范围内,化合物2~9对HBsAg的分泌表达有一定的抑制作用。以上结果均为体外试验结果,还有待进一步进行体内活性研究。
3 结论
以常春藤皂苷元为原料,通过酯化、乙酰化及醚化等反应,合成了新型常春藤皂苷元脂溶性衍生物(2~9),其中3, 4, 6~9未见文献报道。本研究原料来源于贵州地区黄褐毛忍冬的提取物,为开发利用贵州丰富的资源提供参考,还对解决常春藤皂苷元口服不吸收、生物利度低的难题提供依据。此外,在合成时,反应条件温和,产率比较高,脂溶性增大,该方法可以合成较多类型的常春藤皂苷元衍生物。体外抗HBV活性评价分析表明:3~9随浓度的增大对细胞活力的抑制率明显增加,特别是7和9最明显。同时在一定浓度范围内,2~9对HBsAg的分泌表达有一定的抑制作用。
考虑到常春藤皂苷元衍生物在体内的降解过程中还会在不同时段和部位释放出具有药理活性的苷元,将进一步通过动物实验研究其在分析体内药动学及药效学的关系、释药时间及体内代谢情况对活性的影响。该研究结果对进一步研究开发常春藤皂苷元奠定了基础。
[1] 梁宝方,程玉芳,薛天,等. 常春藤皂苷元的抗抑郁药效学研究[J].军事医学,2013,37(4):286-289.
[2] 丁兰,侯茜,徐福春,等. 异叶败酱中三萜化合物常春藤皂苷元对人早幼粒白血病细胞HL-60的增殖抑制、周期阻滞及凋亡诱导作用[J].西北师范大学学报,2009,45(1):88-92.
[3] 王风强. 刺楸皂苷A-常春藤皂苷元单糖链苷类抗癌活性的基本结构[J].国外医药(植物药分册),2002,17(2):71.
[4] 蒋丹. 预知子化学成分及其生物活性研究[D].吉林:东北师范大学,2006:54-60.
[5] 时京珍,刘耕陶.α-常春藤皂甙和无患子皂甙B对小鼠肝微粒体细胞色素P-450的作用[J].中国药理学报,1996,17(3):266.
[6] 孙璐. 维药斯亚旦抗肝癌有效成分研究[D].天津:天津理工大学,2014:45-68.
[7] 王国华. 抗抑郁药物的神经可塑性促进作用及机制研究[D].广东:南方医科大学,2010:55-82.
[8] Chwalek M, Plé K, Voutquenne-Nazabadioko L. Synthesis and hemolytic activity of some hederagenin diglycosides[J].Chem Pharm Bull,2004,52(8):965-971.
[9] 毛玝佳,董登祥,贾宪生,等. 黄褐毛忍冬化学成分及先导物的糖基化研究[J].中国药业,2012,21(22):30-31.
[10] 吴耀民,毛俊琴,李铁军,等. 常春藤皂苷元及衍生物用于制备抗抑郁产品的用途:CN 101214250A[P].2008.
[11] 麻秀萍,贾宪生,韦环. HPLC测定黄褐毛忍冬中的黄褐毛忍冬皂苷甲[J].华西药学杂志,2011,26(2):168-169.
[12] 国家药典委员会. 中华人民共和国药典(一部)[S].北京:中国医药科技出版社,2010:28.
[13] Aokia T, Shido K, Takahashi Y,etal. Structures of 3,28-O-bisglycosidic triterpenoid saponins ofFatsiajaponica[J].Phytochemistry,1981,20(20):1681-1886.
[14] Kizu H, Tomimori T. Studies on the constituents of Clematis species. Ⅳ. On the saponins of the root of Clematis chinensis Osbeck Ⅳ[J].Chem Pharm Bull,1982,28(9):2827-2830.
[15] 韩金玉,肖剑,王华,等. 紫杉醇溶解度的测定与关联[J].化工学报,2001,52(1):65-67.
[16] 毛盾, 唐建华,沈思康,等. 大豆苷元在有机溶剂中溶解度的测定和关联[J].北京化工大学学报(自然科学版),2015,42(5):48-52.
[17] 段新方,张站斌,段新红. 5,3,4′-三羟基,7-二甲氧基黄酮的另法全合成[J].有机化学,2003,23(4):353-355.
Synthesis and In vitro Anti-HBV Activities of Novel Hederagenin Derivatives
HONG Kai-wen1, JIA Xian-sheng2, YANG Xian-jiang2, DONG Deng-xiang2*
(1. Applied Medical Department, Vocational and Technical College of Anshun, Anshun 561000, China; 2. College of Pharmacy, Guiyang College of Traditional Chinese Medicine, Guiyang 550001, China)
Eight derivatives(2~9) of hederagenin were synthesized by acetylation, etherification, benzoylation and esterification,etal, on 3,23-hydroxyl and 28-carboxyl of hederagenin. Among them, six derivatives were not reported. The structures were characterized by1H NMR,13C NMR, IR and MS. The anti-HBV activities of the compounds were determined by MTT method. The results showed that cell viabilities of 3~9 was significantly inhibited with increasing of concentration. In particular, inhibition ratio of 7 and 9 were 9.46±0.47% at concentration of 1×10-6mol·L-1. The inhibition abilities on Hep G2.2.15 cells expressing HBs Ag were tested. The results showed that 2~9 have certain inhibitory effect on the secretory expression of HBsAg.
hederagenin; derivative; synthesis; anti-HBV activity
2016-10-08
贵州省科技攻关项目(黔科合SY【2010】3016号)
洪开文(1987-),男,穿青族,贵州安顺人,硕士研究生,主要从事中药化学与民族药研究。 E-mail: 854374072@qq.com
董登祥,副教授, Tel. 0851-33431699, E-mail: ddx331@163.com
O621.3; O629.2
A
10.15952/j.cnki.cjsc.1005-1511.2017.01.16250
猜你喜欢
吉林农业(2019年6期)2019-06-11
教育教学论坛(2018年38期)2018-09-25
中成药(2018年8期)2018-08-29
现代园艺(2018年3期)2018-02-10
中成药(2017年9期)2017-12-19
中成药(2017年9期)2017-12-19
中成药(2017年10期)2017-11-16
中成药(2017年4期)2017-05-17
医学研究杂志(2015年8期)2015-06-22
特产研究(2014年4期)2014-04-10
