
新型肟基取代豆甾类化合物的合成及其抗肿瘤活性
2017-02-07 08:59甘春芳刘晓兰庞丽萍庞春玲黄燕敏崔建国
合成化学 2017年1期
甘春芳, 董 新, 刘晓兰, 庞丽萍, 庞春玲, 黄燕敏, 崔建国
(广西师范学院 北部湾环境演变与资源利用教育部重点实验室 化学与材料科学学院,广西 南宁 530001)
·研究论文·
新型肟基取代豆甾类化合物的合成及其抗肿瘤活性
甘春芳, 董 新, 刘晓兰, 庞丽萍, 庞春玲, 黄燕敏, 崔建国*
(广西师范学院 北部湾环境演变与资源利用教育部重点实验室 化学与材料科学学院,广西 南宁 530001)
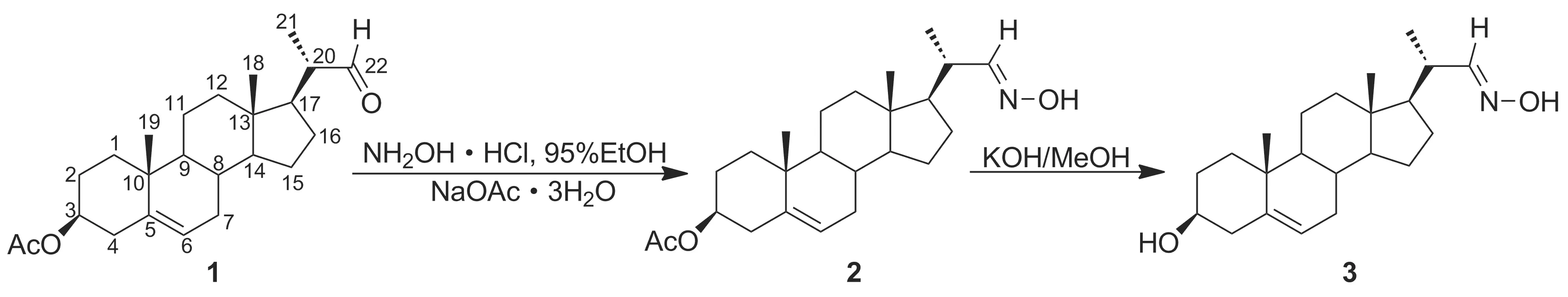
以豆甾醇为原料,通过臭氧化将豆甾醇的C20—C22键断裂,再经过官能团转换,合成了22-肟基取代的单肟基化合物(3和9), 6,22-二肟基取代的双肟基化合物(13和14)及3,6,22-三肟基化合物(17),其中涉及4个中间体(5~8)及目标化合物9, 13, 14和17共8个新化合物,其结构经1H NMR,13C NMR, IR和HR-MS(ESI)表征。采用MTT法测试了化合物对人胃癌细胞(SGC-7901)、人肝癌细胞(Bel-7404)和人体乳腺癌细胞株(HeLa)的体外抗肿瘤活性。结果表明,具有22-肟基取代的3-羟基-5-烯结构的豆甾化合物3对受试细胞均有一定活性,IC50分别为34±2 μmol·L-1, 32±1 μmol·L-1和38±3 μmol·L-1。但是进一步在甾核上引入肟基或羟基的其他几种类型化合物的抗肿瘤活性没有提高。
豆甾醇; 22-肟甾体化合物; 合成; 抗肿瘤增殖活性; 细胞毒性
甾体激素在维持生命、调节机体性能、提高免疫力及促进机体发育等方面具有不可替代的作用,是人类生命的金钥匙。对甾核的结构进行改造,如引入不同的官能团,改变甾核的侧链,或取代甾核的骨架结构,在甾核中引入一个或多个杂原子都有可能引起其生理活性的变化[1-5]。
某些含氮的甾体化合物被用于癌症的治疗,然而由于其对人体组织具有较高毒性而限制了其应用[6-8]。甾体肟类化合物在临床上主要用于雌性激素和避孕等方面,一些肟基取代的化合物具有抗癌、抗菌及抗病毒等生理活性,引起了广大医药研究及化学研究人员的关注[9-15]。近年来,本课题组[16-20]一直致力于甾体抗肿瘤药物的研究,研究发现,当甾体的母核结构为胆甾烷支链结构,甾核的3,6-位被羟基或肟基取代时,某些化合物具有较好的细胞毒性。
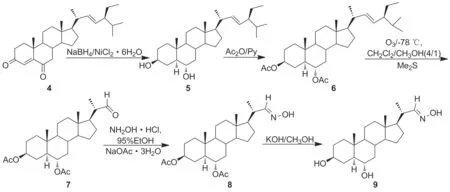
为了寻找高效、低毒的甾体类抗肿瘤化合物,本文设计并合成了4个不同系列的22-肟基取代豆甾化合物。即以豆甾醇为原料,通过臭氧化将豆甾醇的C20—C22键断裂,再经过官能团转换,分别合成了3-羟基,22-肟基和3,6-二羟基,22-肟基的单肟基化合物(3和9), 3-羟基(或乙酰基),6,22-二肟基的双肟基化合物(13和14)及3,6,22-三肟基化合物(17)(Scheme 1),其中涉及4个中间体(5~8)及目标化合物9, 13, 14和17共8个新化合物,其结构经1H NMR,13C NMR, IR和HR-MS(ESI)表征。并对化合物的体外抗肿瘤活性进行筛选,研究其构效关系,为甾体肟类化合物的医药开发应用提供理论和实践基础。
1 实验部分
1.1 仪器与试剂
X6型显微熔点仪(温度未校正);Bruker AV-300型超导核磁共振仪(CDCl3为溶剂,TMS为内标);Thermo Scientific Nicolet IS-10型傅立叶红外光谱仪(KBr压片); Agilent 6210 TOFMS型质谱仪;MLLTISKAN MK3型酶标仪。
1[21], 4, 10, 15[17]参考文献方法合成;RPMI 1640培养基,HyClone公司;MTT,Sigma公司;胎牛血清,浙江天杭生物科技有限公司;青霉素-链霉素溶液(100X),Beyotime公司;胰酶,Sigma公司;DMSO,Amresco公司;肿瘤细胞购自上海通派生物细胞库;豆甾醇及其余所用试剂均为分析纯,百灵威科技有限公司,其中溶剂按常规方法干燥。
1.2 合成
(1) 22-降-3β-乙酰氧基豆甾-5-烯-22-肟(2)的合成
在反应瓶中依次加入1 54 mg(0.15 mmol), 95%乙醇10 mL,于55~60 ℃搅拌使其溶解;加入AcONa·3H2O 24 mg(0.175 mmol),搅拌使其溶解;加入NH2OH·HCl 13 mg(0.18 mmol),于60 ℃反应1 h[TLC跟踪,展开剂:A=V(石油醚) ∶V(乙酸乙酯)=4 ∶1]。减压蒸除大部分乙醇(出现大量白色固体),残余物加少许水(出现白色固体)。用乙酸乙酯(3×10 mL)萃取,合并萃取液,依次用饱和食盐水洗涤,无水硫酸钠干燥,减压蒸出乙酸乙酯得淡黄色固体,经硅胶柱层析(洗脱剂:A=4 ∶1)纯化得2 42 mg,产率75%, m.p. 175~177 ℃;1H NMRδ: 7.28(d,J=8.1 Hz, 1H, 22-H), 7.23(br s, 1H, NOH), 5.39(br d,J=4.8 Hz, 1H, 6-H), 4.67~4.56(m, 1H, 3-Hα), 2.05(s, 3H, CH3CO), 1.14(d,J=6.8 Hz, 3H, 21-H), 1.04(s, 3H, 19-H), 0.74(s, 3H, 18-H);13C NMRδ: 170.6(3-COCH3), 156.9(C22), 139.7(C5), 122.6(C6), 73.9(C3), 56.5(C14), 53.9(C17), 50.0(C9), 42.6(C13), 39.5(C12), 38.1(C4), 37.1(C10), 37.0(C1), 36.6(C20), 31.8(C7), 31.8(C8), 27.7(C2), 27.6(C16), 24.3(C15), 21.4(CH3CO), 21.0(C11), 19.3(C19), 17.9(C21), 12.1(C18); IRν: 3 358(OH), 2 933(CH3), 1 732(C=O), 1 442(C=N) cm-1。
(2) 22E-肟-22,23-断豆甾-5-烯-3β-醇(3)的合成
将2 40 mg(0.103 mmol)溶于1 mol·L-1KOH/CH3OH(10 mL)中,搅拌下于室温反应1 h[TLC跟踪(展开剂:A=2 ∶1)]。减压蒸去溶剂得白色固体,加入冰水10 mL,用乙酸乙酯(3×10 mL)萃取,合并萃取液,依次用水和饱和食盐水洗涤,无水硫酸钠干燥,减压蒸除溶剂得透明油状液体,经硅胶(200~300目)柱层析(洗脱剂:A=2 ∶1)纯化得白色固体3 32 mg,产率90%, m.p. 154~155 ℃;1H NMRδ: 7.28(d,J=7.8 Hz, 1H, 22-H), 5.35(br s, 1H, 6-H), 3.61~3.45(m, 1H, 3-Hα), 1.12(d,J=6.3 Hz, 3H, 21-H), 1.01(s, 3H, 19-H), 0.72(s, 3H, 18-H);


Scheme 1
13C NMRδ: 156.7(C22), 140.7(C5), 121.6(C6), 71.7(C3), 56.5(C14), 53.9(C17), 50.1(C9), 42.6(C13), 42.2(C4), 39.5(C12), 37.2(C1), 37.1(C20), 36.5(C10), 31.9(C8), 31.8(C7), 31.5(C2), 27.7(C16), 24.3(C15), 21.0(C11), 19.4(C19), 17.9(C21), 12.1(C18); IRν: 3 416(OH), 2 933(CH3), 1 640(C=C), 1 454(C=N), 1 053(C—O) cm-1; HR-MS(ESI)m/z: Calcd for C22H36NO2{[M+H]+}346.274 6, found 346.272 7。
(3) 5α-豆甾-22-烯-3β,6α-二醇(5)
将4 839 mg(1.98 mmol)溶于甲醇(50 mL)中,搅拌下加热使其完全溶解;加入NiCl2·6H2O 471 mg(1.98 mmol),溶解后,加入NaBH4451 mg(11.9 mmol),反应1 h(TLC监测,展开剂:A=2 ∶1)。用1 mol·L-1盐酸淬灭反应,蒸出大部分甲醇,用乙酸乙酯(3×20 mL)萃取,合并萃取液,依次用水、饱和碳酸氢钠溶液和饱和食盐水洗涤,无水硫酸钠干燥,减压除去溶剂得白色固体797 mg,经硅胶柱层析(洗脱剂:A=2 ∶1)纯化得白色固体5 613 mg,产率72%,m.p. 204~206 ℃;1H NMRδ: 5.17(dd,J=15.0 Hz, 8.4 Hz, 1H, 23-H), 5.03(dd,J=15.0 Hz, 8.7 Hz, 1H, 22-H), 4.23~4.17(m, 1H, 3-Hα), 3.78~3.74(m, 1H, 6-Hβ), 1.04(d,J=6.3 Hz, 3H, 21-H), 1.03(s, 3H, 19-H), 0.87(d,J=6.3 Hz, 3H, 27-H), 0.82(t,J=6.9 Hz, 3H, 29-H), 0.81(d,J=6.3 Hz, 3H, 26 or 27-H), 0.73(s, 3H, 18-H);13C NMRδ: 138.3(C22), 129.3(C23), 72.3(C3), 66.7(C6), 56.3(C17), 56.1(C14), 54.2(C24), 51.2(C5), 42.6(C13), 41.7(C9), 40.5(C20), 39.8(C12), 39.7(C1), 36.1(C10), 34.0(C7), 33.1(C4), 31.9(C8), 30.3(C25), 29.1(C2), 28.9(C16), 25.4(C28), 24.3(C15), 21.2(C27), 21.1(C26), 20.6(C11), 19.0(C21), 14.9(C19), 12.3(C18), 12.2(C29); IRν: 3 432(OH), 2 933(CH3), 1 640(C=C), 1 037(C—O) cm-1。
(4) 3β,6α-二乙酰氧基-5α-豆甾-22-烯(6)和3β-乙酰氧基-6-氧代豆甾-22-烯(11)的合成(以6为例)
将5 688 mg(1.6 mmol)溶于吡啶(15 mL)中,搅拌使其溶解;缓慢滴加乙酸酐2.5 mL,滴毕,反应0.5 h。于室温静置24 h,加蒸馏水15 mL,用乙酸乙酯(3×10 mL)萃取,依次用1 mol·L-1盐酸、饱和NaHCO3溶液及水洗涤,再用饱和食盐水洗涤至中性,无水硫酸钠干燥,减压蒸出溶剂,当有较大量固体析出时,停止蒸馏,加热使固体溶解,静置结晶得白色片状晶体6 738 mg。
用类似方法合成11。
6: 产率90%, m.p. 120~122 ℃;1H NMRδ: 5.17(dd,J=15.0 Hz, 8.4 Hz, 1H, 22-H), 5.03(dd,J=15.0 Hz, 8.1 Hz, 1H, 22-H), 4.98~4.92(m, 1H, 6-Hβ), 4.79~4.68(m, 1H, 3-Hα), 2.06(s, 3H, 6-COCH3), 2.04(s, 3H, 3-COCH3), 1.03(s, 3H, 19-H), 1.02(d,J=6.0 Hz, 3H, 21-CH3), 0.86(d,J=6.3 Hz, 3H, 26 or 27-H), 0.82(t,J=7.2 Hz, 3H, 29-H), 0.81(d,J=6.3 Hz, 3H, 26 or 27-H), 0.72(s, 3H, 18-H);13C NMRδ: 170.6(6-COCH3), 170.5(3-COCH3), 138.2(C22), 129.4(C23), 73.5(C6), 73.4(C3), 56.1(C17), 56.0(C14), 53.9(C9), 51.2(C24), 46.2(C5), 42.5(C13), 40.5(C20), 39.7(C12), 38.0(C1), 36.3(C10), 35.5(C7), 31.9(C8), 31.3(C2), 31.0(C25), 28.8(C4), 27.3(C16), 25.4(C28), 24.2(C15), 21.4(6-CH3CO), 21.3(3-CH3CO), 21.2(C27), 21.1(C26), 20.9(C11), 19.0(C21), 15.1(C19), 12.3(C18), 12.2(C29); IRν: 2 958(CH3), 1 740(C=O), 1 707(C=O), 1 610(C=C), 1 025(C—O) cm-1。
11: 产率83%, m.p. 138~140 ℃;1H NMRδ: 5.16(dd,J=15.3 Hz, 8.4 Hz, 1H, 23-H), 5.03(dd,J=15.3 Hz, 8.4 Hz, 1H, 22-H), 4.74~4.63(m, 1H, 3-Hα), 2.36~2.25(m, 2H, 7-H), 2.04(s, 3H, 3-CH3CO), 1.03(d,J=6.6 Hz, 3H, 21-H), 0.82(t,J=7.5 Hz, 3H, 26 or 27-H), 0.80(d,J=6.6 Hz, 3H, 26 or 27-H), 0.78(s, 3H, 19-H), 0.69(s, 3H, 18-H);13C NMRδ: 210.4(C6), 170.6(3-COCH3), 138.0(C22), 129.6(C23), 72.8(C3), 56.8(C14), 56.5(C17), 55.9(C5), 53.8(C9), 51.2(C24), 46.7(C7), 42.9(C13), 41.0(C20), 40.4(C12), 39.3(C10), 37.9(C8), 36.4(C1), 31.9(C25), 28.7(C16), 26.8(C2), 26.1(C28), 25.4(C4), 24.0(C15), 21.5(C11), 21.3(3-CH3CO), 21.2(C27), 21.1(C26), 19.0(C21), 13.0(C19), 12.3(C18), 12.2(C29); IRν: 2 958(CH3), 1 740(C=O), 1 707(C=O), 1 466(C=C) cm-1。
(5) 3β,6α-二乙酰氧基-22,23-断-5α-豆甾-22-醛(7), 3β-乙酰氧基-6-氧代-22,23-断-5α-豆甾-22-醛(12)和3,6-二氧代-22,23-断-5α-豆甾-22-醛(16)的合成(以7为例)
将6 340 mg(0.24 mmol)加至圆底烧瓶中,加入二氯甲烷16 mL和甲醇4 mL,搅拌使其完全溶解;置于液氮冷却的乙酸乙酯保温瓶中冷却至-78 ℃,通入富含有O3的氧气流,反应30 min(TLC跟踪,展开剂:A=4 ∶1);停止通入O3,此时溶液变为淡蓝色,通入氧气反应0.5 h;加入二甲硫醚3 mL,升至室温,反应过夜。减压蒸除大部分溶剂得白色固体的混合油状物,用二氯甲烷(50 mL)溶解,加入蒸馏水10 mL,分液,有机层用无水硫酸钠干燥,减压蒸除溶剂得无色透明油状物,干燥后经快速柱层析(洗脱剂:A=4 ∶1)纯化得油状液体7 214 mg。
用类似方法合成12和16。
7: 产率74.7%;1H NMRδ: 9.57(d,J=3.3 Hz, 1H, 22-H), 4.98~4.90(m, 1H, 6-Hβ), 4.78~4.67(m, 1H, 3-Hα), 2.42~2.31(m, 1H, 20-H), 2.05(s, 3H, 6-CH3CO), 2.03(s, 3H, 3-CH3CO), 1.13(d,J=6.6 Hz, 3H, 21-H), 1.03(s, 3H, 19-H), 0.75(s, 3H, 18-H);13C NMRδ: 205.0(C22), 180.9(6-COCH3), 170.6(3-COCH3), 73.4(C6), 73.2(C3), 55.2(C14), 53.8(C9), 51.0(C17), 49.4(C20), 46.1(C5), 43.2(C13), 39.4(C12), 38.0(C1), 36.3(C7), 35.5(C10), 31.0(C8), 30.9(C4), 27.3(C2), 27.0(C16), 24.5(C15), 21.3(6-CH3CO), 21.4(3-CH3CO), 20.9(C11), 15.1(C19), 13.4(C21), 12.5(C18); IRν: 2 941(CH3), 1 736(C=O), 1 728(C=O), 1 707(C=O) cm-1。
12: 无色胶状物,产率71%, m.p. 154~156 ℃;1H NMRδ: 9.60(d,J=3.0 Hz, 1H, 22-CHO), 4.74~4.63(m, 1H, 3-Hα), 2.37~2.26(m, 2H, 7-H), 2.05(s, 3H, 3-CH3CO), 1.15(d,J=6.6 Hz, 3H, 21-H), 0.80(s, 3H, 19-H), 0.73(3H, s, 18-H);13C NMRδ: 210.0(C6), 204.7(C22), 170.6(3-COCH3), 72.8(C3), 56.5(C14), 55.9(C5), 53.8(C9), 50.9(C17), 49.4(C20), 46.5(C7), 43.5(C13), 41.0(C12), 39.1(C10), 37.8(C8), 36.4(C1), 26.9(C2), 26.8(C4), 26.1(C16), 24.3(C15), 21.4(3-CH3CO), 21.3(C11), 13.4(C19), 13.0(C18), 12.4(C21); IRν: 2 937(CH3), 1 723(C=O), 1 707(C=O) cm-1。
16: 白色固体,产率70%, m.p. 168~169 ℃;1H NMR(DMSO-d6)δ: 9.54(d,J=6.5 Hz, 1H, 22-H), 2.78(dd,J=13.5 Hz, 4.0 Hz, 1H, 5-H), 1.05(d,J=6.5 Hz, 3H, 21-H), 0.87(s, 3H, 19-H), 0.69(s, 3H, 18-H);13C NMRδ: 209.4(C3), 209.3(C6), 205.7(C22), 56.5(C14), 55.5(C5), 52.4(C17), 51.0(C20), 49.0(C13), 46.2(C9), 43.5(C7), 41.0(C10), 39.0(C4), 37.7(C12), 37.6(C2), 37.4(C1), 37.2(C8), 26.9(C16), 24.4(C15), 21.6(C11), 13.6(C19), 12.6(C18), 12.5(C21); IRν: 2 931(CH3), 1 709(C=O) cm-1。
(6) 22,23-断-3β,6α-二乙酰氧基-5α-豆甾-22E-肟(8), 3β-乙酰氧基-22,23-断-5α-豆甾-6E,22E-二肟(13)和(3E,6E,22E)-三肟基-22,23-断-5α-豆甾烷(17)的合成(以8为例)
在反应瓶中依次加入7 242 mg(0.56 mmol)和95%乙醇30 mL,加热搅拌使其溶解;加入三水乙酸钠77 mg(0.56 mmol), 溶解后分3批缓慢加入盐酸羟胺49 mg(0.7 mmol),加毕(10 min),溶解完全后,控制反应温度在65 ℃左右反应1 h(TLC跟踪,展开剂:A=4 ∶1)。减压蒸除大部分乙醇(析出大量白色固体),加少许蒸馏水(析出白色沉淀),加入乙酸乙酯溶解,用乙酸乙酯(3×15 mL)萃取,合并萃取液,依次用蒸馏水和饱和食盐水洗涤,无水硫酸钠干燥,减压蒸除溶剂得白色固体,经硅胶柱层析(洗脱剂:A=4 ∶1)纯化得8 207 mg。
用类似方法合成13和17。
8: 白色固体,产率82.7%, m.p. 90~91 ℃;1H NMRδ: 7.56(br s, 1H, NOH), 7.27(d,J=8.1 Hz, 1H, 22-H), 4.95(br d,J=2.7 Hz, 1H, 6-Hβ), 4.78~4.67(m, 1H, 3-Hα), 2.34~2.44(m, 1H, 20-H), 2.06(s, 3H, 6-CH3CO), 2.04(s, 3H, 3-CH3CO), 1.13(d,J=6.6 Hz, 3H, 21-H), 1.03(s, 3H, 19-H), 0.75(s, 3H, 18-H);13C NMRδ: 170.6(3, 6-COCH3), 156.7(C22), 73.4(C6), 73.3(C3), 55.8(C14), 53.9(C17), 53.8(C9), 46.1(C13), 42.9(C5), 39.5(C12), 38.0(C10), 37.1(C1), 36.3(C7), 35.5(C20), 30.9(C8), 29.7(C4), 27.6(C2), 27.3(C16), 24.2(C15), 21.4(6-CH3CO), 20.9(3-CH3CO), 18.4(C11), 17.9(C21), 15.1(C19), 12.4(C18); IRν: 3 452(OH), 2 937(CH3), 1 740(C=O), 1 728(C=O), 1 638(C=C) cm-1。
13: 白色固体,产率92.8%, m.p. 132~133 ℃;1H NMRδ: 8.35(s, 1H, NOH), 7.87(s, 1H, NOH), 7.28(d,J=7.8 Hz, 1H, 22-H), 4.75~4.64(m, 1H, 3-Hα), 3.34(dd,J=13.5 Hz, 4.5 Hz, 1H, 7-Hβ), 2.39(dd,J=15.6 Hz, 7.8 Hz, 1H, 5-H), 2.04(s, 3H, 3-CH3CO), 1.14(d,J=6.6 Hz, 3H, 21-H), 0.79(s, 3H, 19-H), 0.71(s, 3H, 18-H);13C NMRδ: 170.7(3-COCH3), 159.6(C6), 156.5(C22), 73.2(C3), 56.3(C14), 54.2(C17), 53.9(C9), 49.3(C5), 43.2(C13), 39.3(C12), 38.9(C10), 37.2(C20), 36.0(C1), 35.7(C8), 29.5(C4), 27.6(C7), 27.5(C2), 27.0(C16), 24.1(C15), 21.4(3-CH3CO), 21.3(C11), 17.8(C19), 12.5(C21), 12.3(C18); IRν: 3 374(OH), 2 941(CH3), 1 723(C=O), 1 650(C=C), 1 450(C=N) cm-1。
17: 白色固体,产率65%, m.p.173~175 ℃;1H NMR(CD3OD)δ: 7.20(d,J=4.8 Hz, 1H, 22-H), 3.38~3.32(m, 2H, 2-Hα, 7-Hβ), 0.76(s, 3H, 18-H), 1.12(d,J=6.5 Hz, 3H, 21-H), 0.88(s, 3H, 19-H);13C NMR(CD3OD)δ: 159.7(C6), 158.2(C3), 155.7(C22), 56.2(C14), 53.9(C17), 50.9(C9), 49.6(C5), 42.7(C13), 39.3(C12), 39.2(C10), 36.8(C20), 35.5(C8), 29.0(C7), 28.0(C2), 27.2(C1), 26.9(C16), 23.7(C19), 21.1(C15), 20.8(C4), 19.4(C11), 17.1(C21), 11.3(C18); IRν: 3 375(OH), 2 937(CH3), 1 649(C=C), 1 457(C=N) cm-1; HR-MS(ESI)m/z: Calcd for C22H36N3O3{[M+H]+}390.275 7, found 390.274 2。
(7) 22E-肟-22,23-断-5α-豆甾-3β,6α-二醇(9)和6E,22E-二肟基-22,23-断-5α-豆甾-3β-醇(14)的合成(以9为例)
将9 113 mg(0.25 mmol)溶于KOH/CH3OH(5 ∶95)溶液15 mL中,搅拌下于室温反应0.5 h(TLC跟踪,展开剂:A=1 ∶1)。减压蒸除溶剂得白色固体,加冰水10 mL,用乙酸乙酯(3×15 mL)萃取,合并萃取液,依次用水和饱和食盐水洗涤,无水硫酸钠干燥,减压蒸除溶剂得白色固体,经硅胶柱层析(洗脱剂:A=1 ∶1)纯化得白色固体9 75 mg。
用类似方法合成14。
9: 产率82%, m.p. 156~158 ℃;1H NMR(CD3COCD3)δ: 9.44(s, 1H, NOH), 7.20(d,J=7.8 Hz, 1H, 22-H), 3.75(br s, 1H, 6-Hβ), 3.58~3.49(m, 1H, 3-Hα), 2.38~2.27(m, 1H, 20-H), 1.10(d,J=6.6 Hz, 3H, 21-H), 1.06(s, 3H, 19-H), 0.76(s, 3H, 18-H);13C NMR(CD3COCD3)δ: 154.5(C22), 70.9(C3), 70.8(C6), 56.1(C14), 54.5(C17), 54.2(C5), 47.8(C9), 42.7(C13), 40.1(C12), 39.7(C1), 38.6(C7), 37.0(C8), 35.9(C4), 35.4(C10), 31.6(C2), 30.4(C20), 27.5(C16), 24.1(C15), 20.9(C11), 17.6(C19), 15.3(C21), 11.8(C18); IRν: 3 420(OH), 2 933(CH3), 1 695(C=C) cm-1; HR-MS(ESI)m/z: Calcd for C22H38NO3{[M+H]+}364.285 2, found 364.283 3。
14: 白色固体,产率85.5%, m.p. 238~240 ℃;1H NMRδ: 8.27(s, 1H, NOH), 7.55(s, 1H, NOH), 7.28(d,J=7.8 Hz, 1H, 22-H), 3.65~3.55(m, 1H, 3-Hα), 3.34(dd,J=4.5 Hz, 13.8 Hz, 1H, 7-Hβ), 2.45~2.32(m, 1H, 20-H), 1.14(d,J=6.6 Hz, 3H, 21-H), 0.78(s, 3H, 19-H), 0.72(s, 3H, 18-H);13C NMRδ: 159.9(C6), 156.6(C22), 71.1(C3), 56.4(C14), 54.3(C17), 53.9(C9), 49.5(C5), 43.1(C13), 39.4(C12), 38.9(C10), 37.1(C20), 36.2(C1), 35.7(C8), 31.6(C4), 30.8(C2), 29.5(C7), 27.6(C16), 24.1(C15), 21.4(C11), 17.8(C19), 12.6(C21), 12.3(C18); IRν: 3 391(OH), 2 937(CH3), 1 732(C=O), 1 650(C=C), 1 446(C=N) cm-1; HR-MS(ESI)m/z: Calcd for C22H37N2O3{[M+H]+}377.280 4, found 377.278 7。
1.2 抗肿瘤活性测定
采用MTT法测试了化合物对人胃癌细胞(SGC-7901)、人肝癌细胞(Bel-7404)和人体乳腺癌细胞株(HeLa)的体外抗肿瘤活性。
首先将化合物溶解在DMSO中配制成10 mg·mL-1溶液,置冰箱保存;将对数生长期的肿瘤细胞分别以约(1~3)×104个·mL-1的密度接种于96孔板中,每孔接种200 μL,置于CO2培养箱中培养24 h;按预设的浓度梯度加入待测样品,每一浓度梯度设3个平行孔,同时分别采用顺铂作为阳性对照和等量的DMSO作为空白对照;在二氧化碳培养箱中于37 ℃培养72 h。每孔加入MTT(5 mg·mL-1)20 μL,在二氧化碳培养箱中继续温育4 h;抽取上清液,加入DMSO 200 μL,在振动器上震荡10 min溶解沉淀,随后用酶标仪在492 nm波长测定OD值,计算抑制率[抑制率=(1-OD值样品/OD对照)×100%][22],以抑制率对药物浓度作图,求出每个样品的IC50。
2 结果与讨论
2.1 合成
在3的合成中,1与盐酸羟胺反应生成化合物2,随后在KOH的甲醇溶液中进行脱酯反应合成化合物3。
在9的合成中,首先,化合物4在NiCl2存在下用NaBH4还原制得72%的化合物5。然后,通过酯化反应保护3,6-羟基得化合物6。化合物6在4 ∶1的CH2Cl2/MeOH中于-78 ℃进行臭氧化反应,通入O2除去过量O3,并且加入Me2S生成臭氧化物得化合物7。接着,化合物7与盐酸羟胺反应制得化合物8。最后,化合物8在KOH的甲醇溶液中进行脱酯化反应合成化合物9。
为了解多肟基取代对化合物毒性的影响,首先,通过化合物4在Ni2+存在下进行选择性还原制得10,酯化制得化合物11,接着,化合物11进行臭氧化反应生成12。化合物12与盐酸羟胺在乙醇和醋酸钠存在下反应以93%产率获得化合物13。最后,化合物13在KOH/CH3OH(1 mol·L-1)中于室温进行水解反应合成化合物14。
为进一步评价肟基在甾核的不同位置上对化合物细胞毒性的影响,本文还合成了3-, 6-, 22-位三肟基取代的豆甾化合物17。在17的合成中,化合物4还原生成化合物10和5的混合物,然后直接氧化生成豆甾-22-烯-3,6-二酮15,后经臭氧化断键形成醛基,再通过控制盐酸羟胺的用量和反应时间合成化合物17。
在这些合成的化合物中,化合物5, 6, 7, 8, 9, 13, 14, 17均为新化合物。
2.2 抗肿瘤活性
为测定肟基在不同位置对生物活性的影响,本文测试了化合物对SGC 7901, Bel 7404和HeLa肿瘤细胞株的活性,结果见表1。由表1可以看出,具有22-肟基的3-羟基-5-烯结构的豆甾化合物3对受试细胞均有一定的活性,IC50分别为34±2 μmol·L-1, 32±1 μmol·L-1和38±3 μmol·L-1。将3-酯基变为3-羟基时,细胞毒性变化略微提高,然而,将化合物3中的5,6-双键变为6-羟基(9)或6-肟基(14)时,这些化合物的体外抗肿瘤增殖活性降低。但是,在3-, 6-, 22-位同时引入肟基官能团时,具有三肟基取代结构的化合物17的体外抗肿瘤增殖活性与化合物9和14相比有明显的提高。以上结果表明3-肟基官能团在化合物17中对这些肿瘤细胞的毒性有明显的提高。然而,引入3-或(和)6-肟基冠能团并没有明显地提高这些化合物的细胞毒性。从测试的结果与之前我们的工作相比,在甾核中引入多肟基官能团并不能明显提高化合物的细胞毒性。
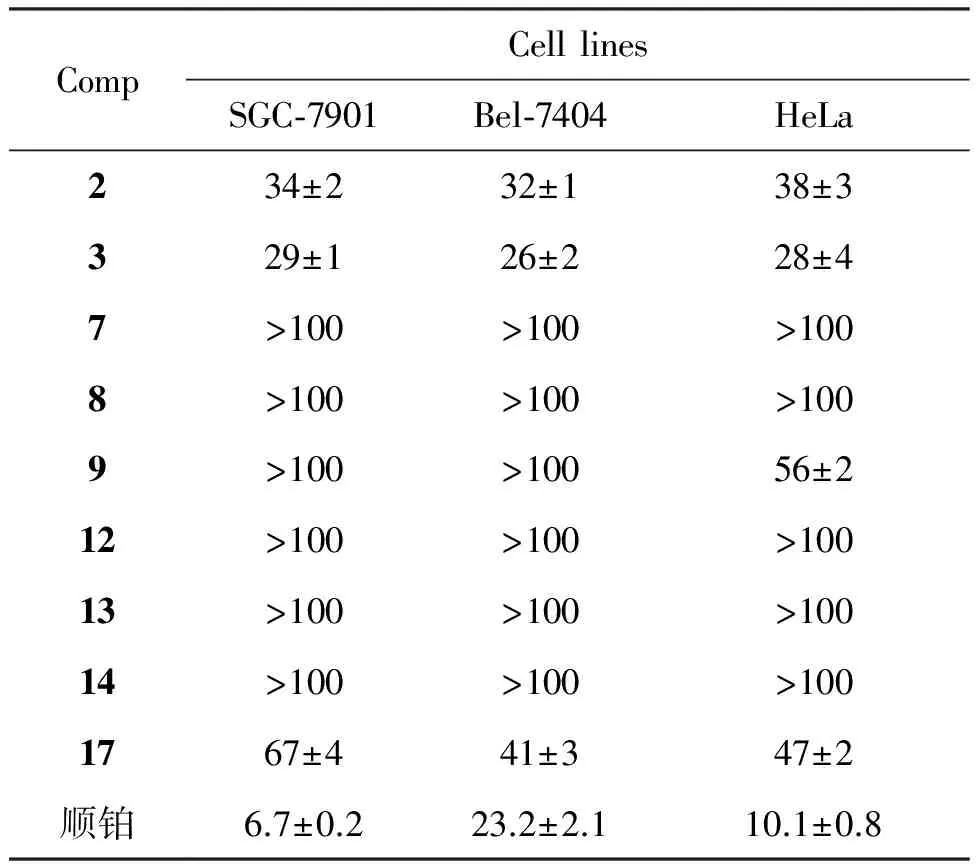
表1 化合物的体外抗肿瘤活性(IC50/μmol·L-1)
以豆甾醇为起始原料,合成了不同系列的22-肟基结构的甾体化合物,包含8个新化合物。其中,具有22-肟基和3-羟基5-烯结构的化合物3对人胃癌细胞(SGC-7901)、人肝癌细胞(Bel-7404)和人体乳腺癌细胞株(HeLa)显示了中等强度的抗肿瘤增殖活性,IC50分别为34±2 μmol·L-1, 32±1 μmol·L-1和38±3 μmol·L-1。然而,在3-或6-位引入肟基时并不能提高这些甾体化合物的抗肿瘤增殖活性。该研究结果为设计新颖的化学抗肿瘤药物提供了理论参考。
[1] Duha C Y, Loa I W, Wang S K,etal. New cytotoxic steroids from the soft coral Clavularia viridis[J].Steroids,2007,72:573-579.
[2] Malika I O, Maurice S. Recent advances in thiasteroids chemistry[J].Steroids,2006,71:1025-1044.
[3] Hanson R. Steroids:Partial synthesis in medicinal chemistry[J].Nat Prod Rep,2006,23:100-107.
[4] 陈思静,崔建国,李莹,等. 具有生理活性甾体腙类化合物的研究进展[J].有机化学,2011,31(2):187-192.
[5] 张晓佳,崔建国,李莹,等. 具有生理活性甾体肟类化合物的研究进展[J].有机化学,2010,30(5):655-661.
[6] 黄燕敏,郑嘉桦,赵丹丹,等. 新型甾体N,N-二甲基缩氨硫脲类化合物的合成及其抗肿瘤活性[J].合成化学,2015,23(12):1106-1110.
[7] Trafalis D T P, Germichalos G D, Koukoulitsa C,etal. Lactandrate:A D-homo-aza-androsterone alkylator in the treatment of breast cancer[J].Breast Cancer Res Treat,2006,97:17-31.
[8] Trafalis D T P. Hybrid aza-steroid alkylators in the treatment of colon cancer[J].Cancer Let,2006,243:202-210.
[9] Koutsourea A I, Fousteris M A, Arsenou E S,etal. Rational design,synthesis,andinvivoevaluation of the antileukemic activity of six new alkylating steroidal esters[J].Bioorg Med Chem,2008,16(9):5207-5215.
[10] Miriam R, Carlos J, Jaime R J. 6E-Hydroximinosteroid homodimerization by cross-metathesis processes[J].Steroids,2007,72:729-735.
[11] Poza J, Rega M, Paz V,etal. Synthesis and evaluation of new 6-hydroximinosteroid analogs as cytotoxic agents[J].Bioorg Med Chem,2007,15:4722-4740.
[12] Khan S A, Asiri A M, Saleem K. Synthesis and biological evaluation of new oxime-ether derivatives of steroid as anti-bacterial agents[J].J Saudi Chem Soc,2012,16(1):7-11.
[13] Bazin M A, Loiseau P M, Letourneux Y,etal. Synthesis of oxysterols and nitrogenous sterols with antileishmanial and trypanocidal activities[J].Europ J Med Chem,2006,41:1109-1116.
[14] Ling Y Z, Li J S, Kato K,etal. Synthesis andinvitroactivity of some epimeric 20 alpha-hydroxy,20-oxime and aziridine pregnene derivatives as inhibitors of human 17 alpha-hydroxylase/C17,20-lyase and 5 alpha-reductase[J].Bioorg Med Chem,1998,6:1683-1693.
[15] Hartmann R W, Hector M, Haidar S,etal. Synthesis and evaluation of novel steroidal oxime inhibitors of P450 17(17α-hydroxylase/C17-20-lyase) and 5α-reductase types 1 and 2[J].J Med Chem,2000,43:4266-4277.
[16] Cui J G, Fan L, Huang L L,etal. Synthesis and evaluation of some steroidal oximes as cytotoxic agents:Structure/activity studies(I)[J].Steroids,2009,74(1):62-72.
[17] Cui J G, Fan L, Huang Y M,etal. Synthesis and evaluation of some steroidal oximes as cytotoxic agents:Structure/activity studies(II)[J].Steroids,2009,74(12):989-995.
[18] Huang Y M, Chen S J, Cui J G,etal. Synthesis and cytotoxicity of A-homo-lactam derivatives of cholic acid and 7-deoxycholic acid[J].Steroids,2011,76:690-694.
[19] Huang Y M, Cui J G, Zhong Z G,etal. Synthesis and cytotoxicity of 17a-aza-d-homo-androster-17-one derivatives[J].Bioorg Med Chem Lett,2011,21:3641-3643.
[20] Gan C F, Fan L H, Huang Y M,etal. Synthesis of novel ring B Abeo-sterol derivatives and their antiproliferative activities[J].Medicinal Chemistry,2013,9:846-854.
[21] Cui J G, Huang L L, Fan L,etal. A facile and efficient synthesis of some (6E)-hydroximino-4-en-3-one steroids,steroidal oximes fromCinachyrellaspp Sponges[J].Steroids,2008,73(3):252-256.
[22] 董见生,袁静梅,何国学,等. 氮杂香豆雌酚衍生物的合成及其抗肿瘤活性[J].合成化学,2016,24(9):774-779.
Synthesis and Antiproliferative Activities of Novel Stigmastane Compounds Substituted with Hydroximino Groups
GAN Chun-fang, DONG Xin, LIU Xiao-lan, PANG Li-ping, PANG Chun-ling, HUANG Yan-min, CUI Jian-guo*
(College of Chemistry and Material Science, Key Laboratory of Beibu Gulf Environment Change and Resources Utilization, Guangxi Teachers Education University, Nanning 530001, China)
A series of stigmastane compounds substituted with 22-hydroximino(3, 9), 6,22-dihydroximino(13, 14) and 3,6,22-trihydroximino(17) were synthesized through ozonation and functional group transformation methods using stigmasterol as starting material. Eight novel compounds including four intermediates(5~8) and target compounds(9, 13, 14 and 17) were involved. The structures were characterized by1H NMR,13C NMR, IR and HR-MS(ESI). The antiproliferative activities of compounds were evaluated against human gastric carcinoma(SGC-7901), human cervical carcinoma(HeLa) and human liver carcinoma(Bel-7404) cells. The results showed that compound 3 with 22-hydroximino and 3-hydroxy-5-enekey features displayed distinct antiproliferative activities to these cells with IC50of 34±2 μmol·L-1, 32±1 μmol·L-1and 38±3 μmol·L-1, respectively, and a further introduction of hydroximino or hydroxyl groupon steroidal nucleus did not enhance the antiproliferative activities of compounds.
stigmasterol; 22-hydroximino groupsteroid; synthesis; antiproliferative activity; cytotoxicity
2016-09-21
国家自然科学基金资助项目(21462009, 21562007); 广西高校科学研究项目(KY2015ZD077)
甘春芳(1973-),女,汉族,广西南宁人,博士,主要从事甾体化合物的合成及抗肿瘤活性研究。 Tel. 0771-3113518,E-mail: ganchunfang2008@126.com
崔建国,教授, Tel. 0771-3908065, E-mail: cuijg1954@126.com
O621.3; O629.2
A
10.15952/j.cnki.cjsc.1005-1511.2017.01.16244
猜你喜欢
城市道桥与防洪(2022年3期)2022-05-08
昆明医科大学学报(2021年8期)2021-08-13
云南化工(2020年11期)2021-01-14
应用化工(2020年9期)2020-09-29
海洋通报(2020年2期)2020-09-04
天然产物研究与开发(2018年7期)2018-08-21
中成药(2018年6期)2018-07-11
天然产物研究与开发(2018年5期)2018-06-13
中成药(2017年8期)2017-11-22
中成药(2017年4期)2017-05-17
