
拟南芥ago1-27突变体的RNA-seq分析
2017-01-19 03:02李盛本莫蓓莘曹晓风陈雪梅
深圳大学学报(理工版) 2017年1期
马 轩,李盛本,莫蓓莘,曹晓风,陈雪梅,4
1)深圳大学生命与海洋科学学院,广东深圳518060;2)中国科学院遗传与发育生物学研究所,北京100101;3)中国农业科学院深圳农业基因组研究所,广东深圳518120;4)美国加利福尼亚大学河滨分校植物科学系, 加利福尼亚河滨CA 92521,美国
【生物工程 / Bioengineering】

拟南芥ago1-27突变体的RNA-seq分析
马 轩1,2,李盛本3,莫蓓莘1,曹晓风2,陈雪梅1,4
1)深圳大学生命与海洋科学学院,广东深圳518060;2)中国科学院遗传与发育生物学研究所,北京100101;3)中国农业科学院深圳农业基因组研究所,广东深圳518120;4)美国加利福尼亚大学河滨分校植物科学系, 加利福尼亚河滨CA 92521,美国
miRNA在植物生长发育和环境适应等方面起着极为重要的作用.本研究对拟南芥ago1-27突变体进行了RNA-seq,鉴定到几千个差异表达基因,并且miRNA靶基因倾向于在ago1-27中表达上调.ago1-27 RNA-seq结合降解组分析鉴定到新的miRNA靶基因,如miR396靶向半胱氨酸蛋白酶基因和miR167靶向编码AT-hook DNA结合蛋白的基因等.
植物生物化学;miRNA;ago1-27基因;RNA-seq;降解组;靶基因;拟南芥
小核糖核酸(microribonucleic acid, microRNA或miRNA)是一类长度约为20~24个核苷酸的内源非编码RNA.miRNA与Argonaute(AGO)蛋白相结合,对靶RNA进行RNA剪切或翻译抑制[1-4].植物miRNA在植物形态建成、生长发育和环境适应等方面起着极为重要的作用[3-4].自2002年首次证实植物中存在miRNA[5]以来,关于植物miRNA的研究进展异常迅速.植物miRNA和靶RNA之间互补紧密,因此,miRNA的靶基因预测较为容易.模式匹配程序scan_for_matches最早被应用于植物miRNA靶基因预测[6].之后,以序列匹配程序FASTA为基础的工具,如Targetfinder和psRNATarget能够快速预测植物靶基因[7-8].降解组(degradome)测序使miRNA靶基因的大规模鉴定成为可能,目前已广泛用于各种植物miRNA靶基因的鉴定中[9-10].
目前已经确认的miRNA靶基因,主要是互补紧密的类型,并集中在高度保守的miRNA上.本研究对模式植物拟南芥的ago1-27突变体进行了RNA-seq,得到了新的候选miRNA靶基因.结合降解组测序数据分析,鉴定出新的miRNA靶基因.
1 材料与方法
1.1 RNA-seq
拟南芥野生型(wild type, WT)和ago1-27突变体的花序用于RNA-seq.使用TRIzol(Invitrogen)提取花序的总RNA.每个样品制备3个生物学重复.使用TruSeq试剂盒(Illumina)构建RNA-seq文库.在IlluminaHiSeq 2500平台上进行50-nt单端非链特异性RNA-seq.原始测序数据已经提交至NCBI SRA数据库(数据号GSE77211).
1.2 生物信息学分析
使用Tophat(v2.0.11,默认参数)将RNA-seq数据与拟南芥基因组(TAIR10)比对,唯一定位的读段(reads)用于后续的分析.应用Cuffdiff进行基因表达分析,采取的标准为:① FDR(false discovery rate)<0.05;② 基因差异表达>2倍.拟南芥miRNA数据源于miRBase(v21),已验证的miRNA靶基因数据源于TarBase(v6.0).miRNA靶基因预测应用TarHunter-L(https://github.com/XMaBio/TarHunter-L).降解组数据应用CleaveLand4进行分析,采用的标准是:① 0类或1类;② 罚分<5.0;③ 剪切位点reads丰度≥10×TP10M,TP10M(transcripts per ten million)为剪切位点reads数占每千万总reads数的比例.
2 结果分析
2.1 拟南芥ago1-27 RNA-seq
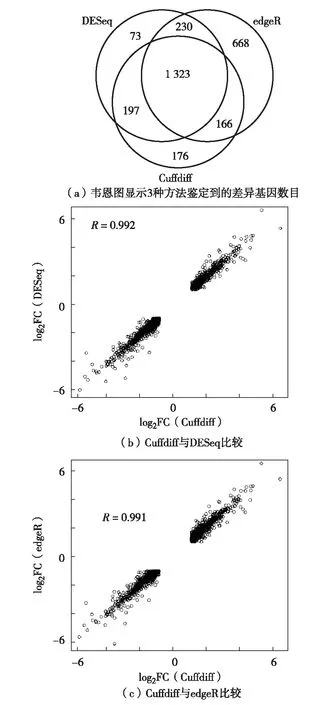
图1 三种RNA-seq分析流程比较Fig.1 Comparison of three RNA-seq analysis methods
对拟南芥野生型和ago1-27突变体植物进行RNA-seq,每个样品进行3个生物学重复.首先比较3种分析RNA-seq数据的流程方法:Tophat2-Cuffdiff、Tophat2-HTSeq-edgeR和Tophat2-HTSeq-DESeq.采用的标准是:① FDR(或调整的P值)<0.05;② 基因差异表达>2.0.Cuffdiff、DESeq和edgeR 3种流程分别鉴定到1 862、1 823和2 387 个差异表达基因,大多数差异表达基因都能被3种方法检测到,如图1(a).其中,edgeR具有较高的灵敏度,DESeq具有最高的特异性.对这3种方法进行两两比较,基因差异表达的倍数(FC为ago1和WT基因差异表达倍数)变化相关性很高(Pearson相关系数> 0.99),如图1(b)和(c),说明该分析方法可靠.因为Cuffdiff方法平衡了灵敏性和特异性,以下结果将基于Cuffdiff的分析方法.同时对3个生物重复进行了比较,3个生物学样品具有高度可重复性(Pearson相关系数> 0.996,如图2(a)和(b),这充分说明了分析样品的可靠性.

图2 三个生物学重复的基因表达谱比较和相关性分析Fig.2 Global gene expression profiles and correlations of three biological replicates
2.2 miRNA靶基因在ago1-27中上调
观察到在ago1-27突变体中AGO1基因的表达值升高,如图3,RPKM(reads per kilobases per million reads)为基因表达值单位.对于这一现象,笔者的解释是:首先, AGO1是miR168的靶基因;在ago1-27突变体中,miRNA与内质网的关联程度下降[11],而内质网膜是miRNA作用的重要场所[11-12],因此在ago1-27中miRNA靶基因的表达失调.应用Cuffdiff在ago1-27突变体中鉴定到689个上调基因和1 175个下调基因,上调和下调的基因中包含24和5个已经验证的miRNA靶基因.这个结果显示,已经确认的miRNA靶基因倾向于富集在ago1-27上调的基因中(Fisher’s exact test,P值= 6.97×10-7).

图3 AGO1基因在ago1-27和野生型中的表达Fig.3 AGO1 gene expressions in ago1-27 and WT
2.3ago1-27 RNA-seq结合降解组分析鉴定新的miRNA靶基因
应用TarHunter-L(见第1章“材料与方法”)预测拟南芥miRNA在编码序列上的靶位点.如图4(a),罚分≤3.0的靶基因在ago1-27突变体中的表达比在野生型的高,而非靶基因在ago1-27中的表达与野生型相当.在ago1-27中上调的预测的靶基因(罚分≤3)中,17个没有报道过(表1).这17个基因包括5个miR396靶基因,还有一些保守miRNA(如miR172和miR167等)的靶基因.

图4 ago1-27 RNA-seq结合降解组鉴定到新的miRNA靶基因Fig.4 ago1-27 RNA-seq combined with degradome analysis identified novel miRNA targets
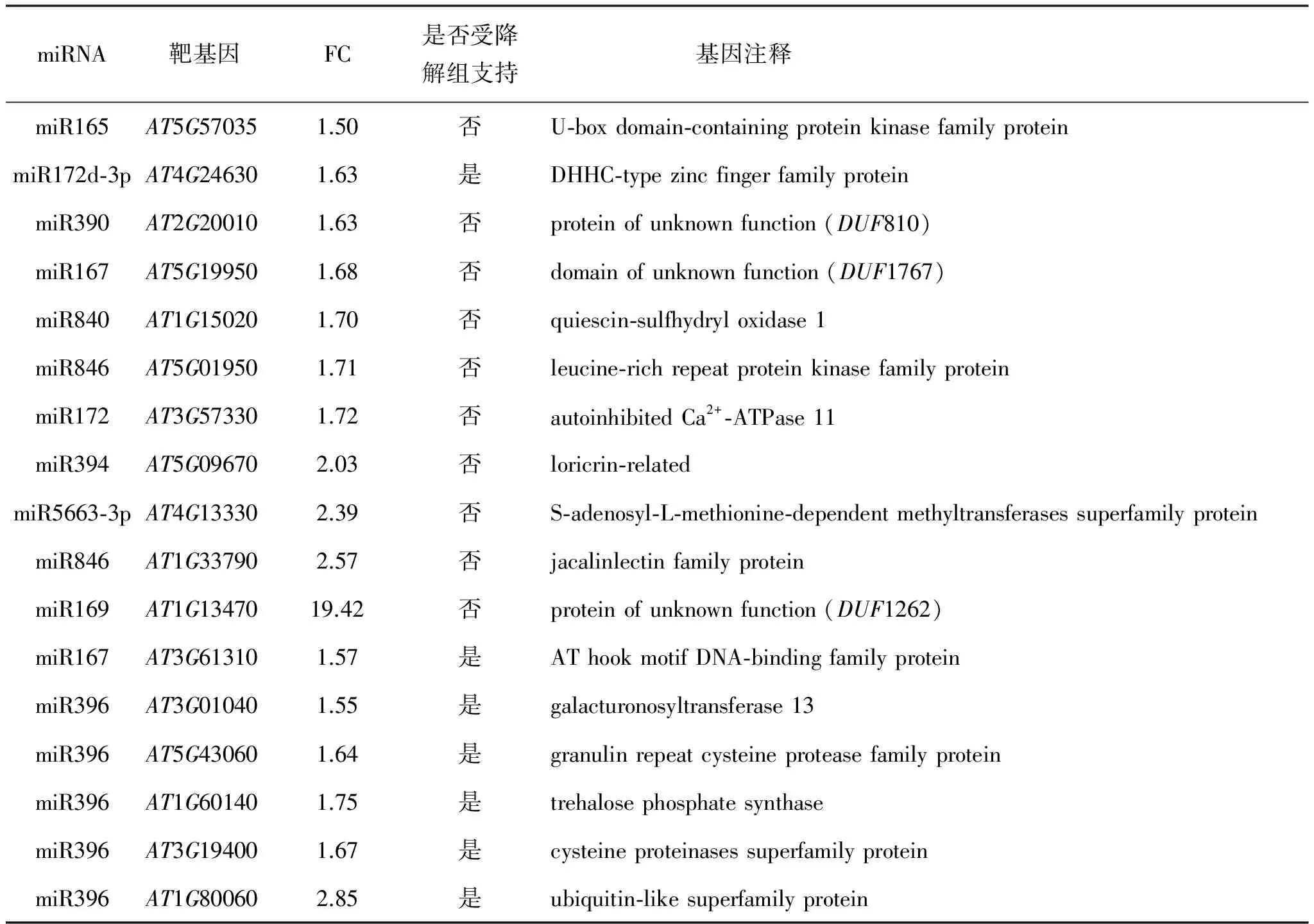
miRNA靶基因FC是否受降解组支持 基因注释miR165AT5G570351.50否U⁃boxdomain⁃containingproteinkinasefamilyproteinmiR172d⁃3pAT4G246301.63是DHHC⁃typezincfingerfamilyproteinmiR390AT2G200101.63否proteinofunknownfunction(DUF810)miR167AT5G199501.68否domainofunknownfunction(DUF1767)miR840AT1G150201.70否quiescin⁃sulfhydryloxidase1miR846AT5G019501.71否leucine⁃richrepeatproteinkinasefamilyproteinmiR172AT3G573301.72否autoinhibitedCa2+⁃ATPase11miR394AT5G096702.03否loricrin⁃relatedmiR5663⁃3pAT4G133302.39否S⁃adenosyl⁃L⁃methionine⁃dependentmethyltransferasessuperfamilyproteinmiR846AT1G337902.57否jacalinlectinfamilyproteinmiR169AT1G1347019.42否proteinofunknownfunction(DUF1262)miR167AT3G613101.57是AThookmotifDNA⁃bindingfamilyproteinmiR396AT3G010401.55是galacturonosyltransferase13miR396AT5G430601.64是granulinrepeatcysteineproteasefamilyproteinmiR396AT1G601401.75是trehalosephosphatesynthasemiR396AT3G194001.67是cysteineproteinasessuperfamilyproteinmiR396AT1G800602.85是ubiquitin⁃likesuperfamilyprotein
为进一步确认通过ago1-27 RNA-seq分析得到的靶基因,对已发表的降解组数据(包括花序和叶片等4个测序文库)进行了分析,鉴定到22个miR396靶基因,包括一些新的靶基因,如FLU、LSU2、RD21B和AT4G32250等.miRNA在靶位点的剪切效率以TP10M表示.在这22个miR396靶基因中,5个没有在ago1-27 RNA-seq中检测到,其余17个基因中的13个在ago1-27突变体中上调变化> 1.5倍,因此,ago1-27 RNA-seq结合降解组分析能有效鉴定miRNA靶基因.
通过ago1-27 RNA-seq分析,还鉴定到AHL11(AT3G61310)是miR167潜在的靶基因,因为它在ago1-27中的表达上调1.6倍.对降解组数据的分析显示,AHL11确实是miR167的靶基因,如图4(b).另外,鉴定到一个果胶甲酯酶(pectin methylesterase,PME)基因AT5G64640是miR156潜在的靶基因,因为它在ago1-27突变体中上调1.6倍,并且受4个降解组数据(成叶、幼叶、野生型花序和xrn4花序)的支持,如图4(c). AT5G64640被划分为A组PME,其在长角果中高度或独特地表达; 该结果说明,miR156除调节SQUAMOSA PROMOTER(SPL)外,还具有调控其他基因的新功能.
3 讨 论
本研究对ago1-27突变体进行了RNA-seq,发现许多已确认的miRNA靶基因在ago1-27中表达上调.以往的研究对少数几个miRNA靶基因进行了分析,在RNA水平它们在ago1-27中表达上调;在蛋白水平它们在ago1-27中上调更为明显[13].因此,ago1-27点突变可能主要影响了miRNA介导的翻译抑制过程.文献[11]的研究表明,在ago1-27中miRNA与内质网膜的关联度下降.由于内质网膜是蛋白翻译的重要场所[14-16],miRNA与膜的解离可能导致miRNA介导的翻译抑制过程的失调.
目前已确认的miRNA靶基因集中在保守miRNA和紧密互补的类型上[17],新的证据表明松弛配对类型的靶基因确实存在[18-20].本研究结合miRNA靶基因预测、降解组数据和ago1突变体RNA-seq分析,鉴定出新的miRNA靶基因,这有助于深入认识植物miRNA和靶基因的调控网络.
/ References:
[1] Bartel D P.MicroRNAs: genomics, biogenesis, mechanism, and function[J].Cell,2004,116(2):281-297.
[2] Rogers K, Chen Xuemei. Biogenesis, turnover, and mode of action of plant microRNAs[J]. The Plant Cell, 2013, 25(7): 2383-2399.
[3] Voinnet O. Origin, biogenesis, and activity of plant microRNAs[J].Cell,2009,136(4):669-687.
[4] Wu Gang.Plant MicroRNAs and development[J].Journal of Genetics and Genomics,2013,40(5):217-230.
[5] Rhoades M W, Reinhart B J, Lim L P, et al. Prediction of plant microRNA targets[J].Cell, 2002,110(4):513-520.
[6] Jones-Rhoades M W, Bartel D P.Computational identification of plant microRNAs and their targets, including a stress-induced miRNA[J].Molecular Cell,2004,14(6):787-799.
[7] Allen E, Xie Zhixin, Gustafson A M, et al. MicroRNA-directed phasing during trans-acting siRNA biogenesis in plants[J].Cell,2005,121(2):207-221.
[8] Dai Xinbin, Zhao P X. psRNATarget: a plant small RNA target analysis server[J]. Nucleic Acids Research, 2011,39(S2):W155-W159.
[9] Addo-Quaye C, Eshoo T W,Bartel D P, et al. Endogenous siRNA and miRNA targets identified by sequencing of the Arabidopsis degradome[J]. Current Biology, 2008,18(10):758-762.
[10] German M A, Pillay M, Jeong D H,et al.Global identification of microRNA-target RNA pairs by parallel analysis of RNA ends[J].Nature Biotechnology,2008,26(8):941-946.
[11] Li Shengben, Le B, Ma Xuan, et al. Biogenesis of phased siRNAs on membrane-bound polysomes in Arabidopsis[J/OL]. ELife,2016,5:e22750. (2016-12-12)[2016-12-20]. https://elifesciences.org/content/5/e22750.doi:10.7554/elife.22750.
[12] Li Shengben, Liu Lin, Zhuang Xiaohong, et al.MicroRNAs inhibit the translation of target mRNAs on the endoplasmic reticulum in Arabidopsis[J].Cell,2013,153(3):562-574.
[13] Brodersen P, Sakvarelidze-Achard L,Bruun-Rasmussen M, et al. Widespread translational inhibition by plant miRNAs and siRNAs[J].Science,2008,320(5880):1185-1190.
[14] Reid D W, Nicchitta C V. Diversity and selectivity in mRNA translation on the endoplasmic reticulum[J]. Nature Reviews Neuroscience, 2015, 16(4):221-231.
[15] Rabouille C. Pathways of unconventional protein secretion[J/OL]. Trends Cell Biology, 2016, in press. (2016-12-16)[2016-12-20]. http://dx.doi.org/10.1016/j.tcb.2016.11.007.
[16] Wang Pengwei, Hawes C, Hussey P J. Plant endoplasmic reticulum-plasma membrane contact sites[J/OL].Trends in Plant Science, 2016, in press. (2016-12-09)[2016-12-20]. http://dx.doi.org/10.1016/j.tplants.2016.11.008.
[17] Cuperus J T, Fahlgren N, Carrington J C. Evolution and functional diversification of miRNA genes[J]. Plant Cell, 2011, 23(2):431-442.
[18] Brousse C, Liu Qikun, Beauclair L, et al. A non-canonical plant microRNA target site[J]. Nucleic Acids Research, 2014, 42(8):5270-5279.
[19] Creasey K M, Zhai Jixian, Borges F, et al. miRNAs trigger widespread epigenetically activated siRNAs from transposons in Arabidopsis[J]. Nature, 2014, 508(7496):411-415.
[20] Wang Feng, Polydore S, Axtell M J. More than meets the eye? Factors that affect target selection by plant miRNAs and heterochromatic siRNAs[J],Current Opinion Plant Biology, 2015,27:118-124.
【中文责编:晨 兮;英文责编:艾 琳】
RNA-seq analysis onArabidopsisago1-27 mutant
Ma Xuan1,2, Li Shengben3, Mo Beixin1, Cao Xiaofeng2, and Chen Xuemei1,4†
1)College of Life Sciences and Oceanography, Shenzhen University, Shenzhen 518060, Guangdong Province, P.R.China 2)Institute of Genetics and Developmental Biology, Chinese Academy of Sciences,Beijing 100101, P.R.China 3)Shenzhen Institute of Agricultural Genomics, Chinese Academy of Agricultural Sciences,Shenzhen 518120, Guangdong Province, P.R.China 4)Department of Botany and Plant Sciences, Institute of Integrative Genome Biology, University of California, Riverside, California 92521, USA
miRNAs play important roles in plant development and adaptation to environment. We conduct the RNA-seq of Arabidopsisago1-27 mutant and has identified thousands of differentially expressed genes. The study shows that miRNA targets tend to be upregulated in theago1-27 mutant. New miRNA targets are identified byago1-27 RNA-seq combined with degradome analysis. For examples, miR396 targets cysteine proteinase genes and miR167 targets a gene encoding AT-hook DNA-binding protein.
phytobiochemistry; microribonucleic acid (miRNA);ago1-27; RNA-seq; degradome; target genes;Arabidopsis
Received:2016-12-23;Accepted:2016-12-27
Foundation:National Natural Science Foundation of China (31571332, 31210103901, 91440105)
† Corresponding author:Academician of American Academy of Sciences, Professor Chen Xuemei. E-mail: xuemei.chen@ucr.edu
:Ma Xuan, Li Shengben, Mo Beixin, et al. RNA-seq analysis onArabidopsisago1-27 mutant[J]. Journal of Shenzhen University Science and Engineering, 2017, 34(1): 27-32.(in Chinese)
Q 946.2
A
10.3724/SP.J.1249.2017.01027
国家自然科学基金资助项目(31571332,31210103901,91440105)
马 轩(1978—),男,深圳大学博士后研究人员. 研究方向:植物miRNA介导的翻译抑制机理. E-mail: xuanma@genetics.ac.cn
引 文:马 轩,李盛本,莫蓓莘,等. 拟南芥ago1-27突变体的RNA-seq分析[J]. 深圳大学学报理工版,2017,34(1):27-32.
猜你喜欢
表面工程与再制造(2022年1期)2022-05-25
深圳大学学报(理工版)(2021年6期)2021-11-29
深圳大学学报(理工版)(2020年6期)2020-11-14
考试与评价·高二版(2020年5期)2020-09-10
现代装饰(2020年4期)2020-05-20
现代装饰(2020年2期)2020-03-03
生命科学研究(2018年1期)2018-05-29
上海农业学报(2017年3期)2017-04-10
安徽医科大学学报(2016年12期)2017-01-15
新课程研究(2016年21期)2016-02-28
