
2,5-二甲基-1,3,4-噻二唑激发态结构动力学的共振拉曼光谱研究
2017-01-18 08:35庞素梅赵彦英
浙江理工大学学报(自然科学版) 2017年1期
庞素梅,赵彦英
(浙江理工大学理学院化学系,杭州 310018)
2,5-二甲基-1,3,4-噻二唑激发态结构动力学的共振拉曼光谱研究
庞素梅,赵彦英
(浙江理工大学理学院化学系,杭州 310018)
获得了2,5-二甲基-1,3,4-噻二唑(DMETD)在乙腈、甲醇和水中的紫外吸收光谱以及三种溶剂中不同激发波长下的共振拉曼光谱,并结合含时密度泛函TD-B3LYP/6-311++G(d,p)计算对DMETD的短时结构动力学进行了研究。通过固态的FT-Raman、FT-IR光谱和DFT计算指认了DMETD在溶剂中的共振拉曼光谱,并对DMETD共振拉曼光谱的强度模式进行了分析。结果表明DMETD在气相和溶剂中的跃迁主体都是π →π*。溶剂模型计算发现溶剂的极性对拉曼强度无影响。这样,DMETD在Franck-Condon区域活性振动模大致可以指认为7个基频v8,v12,v15,v17,v22,v23和 v27以及它们的倍频和组合频,C=N和N-N伸缩振动占据了对拉曼强度的主要部分,说明S2激发态结构动力学也主要沿这两个坐标进行。
2,5-二甲基-1,3,4-噻二唑;紫外光谱;共振拉曼光谱;密度泛函理论计算;激发态结构动力学
0 引 言
1,3,4-噻二唑是一类典型的含N、S原子的五元杂环化合物,其电子结构体系满足4n+2的π电子规则,具有共轭性和芳香性,同时环具有反磁性,因而易发生取代反应[1]。最常见的取代为2,5位取代,而且陆续发现不同种类2,5位取代-1,3,4-噻二唑作为医药中间体显示了很强的生物活性,例如可用作镇痛和抗炎性[2]、抗菌性和抗真菌性[3]、抗抑郁性[4]、抗癌性[5]、抗痨作用[6]和抗氧化性[7]药物等。
2,5位取代噻二唑可以与金属络合,吸附在金属表面,起到缓蚀作用。典型的2,5位取代化合物是2,5-二巯基-1,3,4-噻二唑(DMTD),由于其特殊结构也是过渡金属离子很好的配体[8]。DMTD是一个强的质子给体,它的巯基可以电离出H+,化学吸附在金属表面,将Cu氧化为Cu+,形成-N=C-S-Cu键,另一边硫原子形成配位键H-N-C→S,构成一层结实的单层链状聚合物膜,起到很好的缓蚀效果[9]。2,5位巯基取代会发生质子迁移反应,会以硫醇或硫酮的结构存在,容易与溶剂形成强的氢键网络体系。此外,Rostkowska[10]和他的同事已经采用低温基质隔离技术对5-甲硫基-1,3,4-噻二唑-2-硫酮的互变异构机理进行了研究,结果发现其在气相中以硫酮式结构稳定存在;武海顺等[11]也对5-氨基-1,3,4-噻二唑-2-硫酮的四种互变异构体进行了研究及光谱指认。本文主要采用共振拉曼光谱实验结合DFT计算对2,5位被相同甲基取代后的2,5-二甲基-1,3,4-噻二唑的激发态结构动力学进行研究。
1 实验部分
2,5-二甲基-1,3,4-噻二唑(2,5-Dimethyl-1,3,4-thiadiazole),纯度98%,分子式:C4H6N2S,相对分子质量:133.20,东京化成工业株式会社公司,日本;高纯水,分析纯,99.0%,自制;乙腈,光谱纯,99.9%,TEDIA公司;甲醇,光谱纯,99.9%,天津市永大化学试剂有限公司。
1.2 实验仪器
UV-2501PC紫外可见光谱仪(美国Varian公司);Nicolet Avatar 370傅立叶变换红外光谱仪(美国Termo公司);Nicolet FT-Raman 960傅立叶变换拉曼光谱仪(美国Termo公司);共振拉曼光谱仪(自制)。
1.3 实验方法
根据DMETD在不同溶剂中的紫外吸收光谱实验来确定共振拉曼实验所需的激发波长范围。
分别选取乙腈、甲醇和水作为溶剂,配制DMETD的不同溶液。共振拉曼光谱实验方法已在文献[12-14]中详细描述。通过四倍频激光线(波长266 nm)和氢气受激拉曼位移管获得不同的激发波长。本实验所采用的激发波长为217.8、223.1、228.7、239.5 nm和245.9 nm。在锥形瓶中采用蠕动泵循环进样。拉曼信号的采集是通过激光束照射液膜的背向散射几何结构实现的,而拉曼信号的检测是由CCD检测器实现的。每张谱图是根据样品的信号比来确定取谱时间,大概为60~150s作为一次输出数据。每个样品采集30-90次,叠加得到样品的共振拉曼光谱图。采用Origin软件包对溶液中共振拉曼光谱溶剂峰进行扣减,最后得到样品在溶剂中的共振拉曼光谱图[15]。实验装置中信号收集系统的强度校正原理及共振拉曼光谱的自吸收校正原理参考文献[16]。
2 理论计算
本文分子气相的基态几何结构的优化、振动频率和溶剂化模型的理论计算都是在B3LYP/6-311++G(d,p)水平下获得的。并采用TD-DFT对分子的激发态电子跃迁能和振子强度等信息进行理论计算。本文所有的理论的计算都是采用Gaussian 09W[17]量子化学计算程序包实现的。
3 结果与讨论
3.1 基态几何结构
在我国旅游业民族体育旅游尚处于新兴旅游项目之一。在进行旅游产品设计时,应依托现有民族体育资源,以娱乐休闲为主要目标对民族体育进行宣传,从而使民族体育得到合理的项目设计。高端定制式的民族体育具有一定的针对性,属于小众的体育旅游,只有具有一定经济能力的人才有意愿选择消费。但高端定制民族体育产品也具有较大的弹性,这些特定的消费人群能够根据自身情况对活动项目进行调整。因此,高端定制民族体育项目发展不能拘泥于传统旅游产品发展模式,而要细致分析消费者心理,致力于满足多样化的消费者需求。
2,5-二甲基-1,3,4-噻二唑(DMETD)的2,5位被相同的甲基取代,从而使DMETD分子的结构对称性增大。如图1所示,在B3LYP/6-311++G(d,p)水平下对DMETD几何结构进行优化,计算得到的DMETD为C2v点群,能量为HF=-663.81042664a.u。
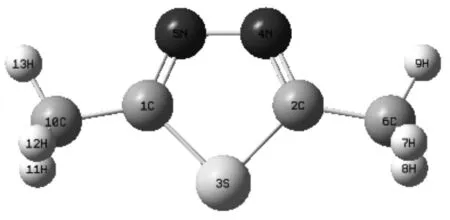
图1 采用B3LYP/6-311++G(d,p)计算的DMETD几何结构优化图
3.2 紫外光谱的研究
DMETD在乙腈、甲醇和水中的紫外吸收光谱图见图2,从图中可以看到DMETD在3种溶剂中的最大吸收位置没有变化,均为217.8 nm,也就是说DMETD在溶剂中的构型与溶剂极性的大小无关。共振拉曼实验所需要的激发波长已经在图中用箭头标示,分别为217.8、223.1、228.4、239.5 nm和245.9 nm。实验测得的DMETD在水、甲醇和乙腈中紫外吸收的摩尔消光系数为εH2O=3909.79 L·mol-1· cm-1,εCH3OH=4566.48 L·mol-1· cm-1,εCH3CN=3456.99 L·mol-1· cm-1。DMETD在气相和溶剂中的电子跃迁能、跃迁轨道和振子强度的计算都是在B3LYP/6-311++G(d, p) 水平下获得的,由于计算结果显示DMETD在3种溶剂模型中的电子跃迁能,电子轨道和振子强度非常接近,因此表中选乙腈的理论计算结果作为对比见表1。而在溶剂中实验的振子强度fH2O=0.1547,fCH3OH=0.1475,fCH3CN=0.1303与气相计算的f=0.1564非常接近。
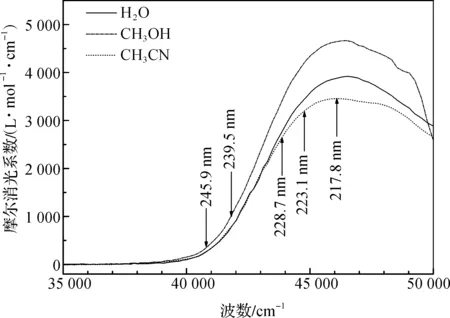
图2 DMETD在水、甲醇和乙腈中的紫外光谱图注:箭头所指为共振拉曼所需的激发波长。
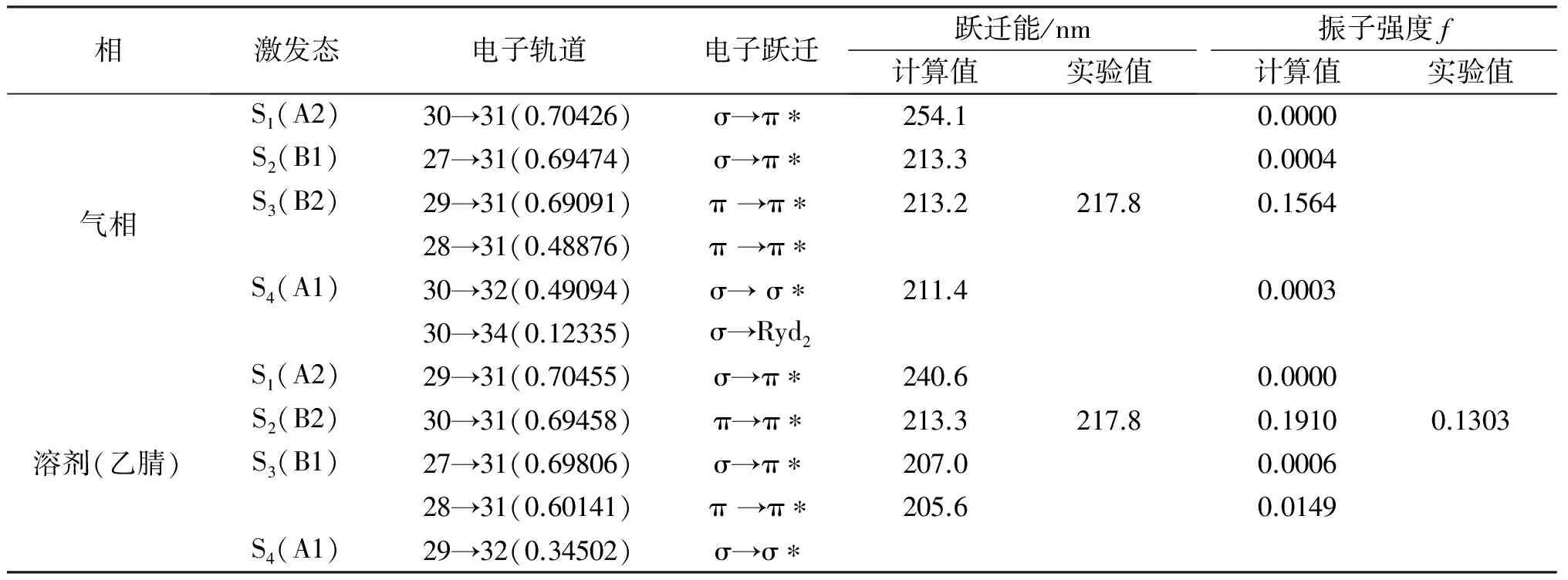
相激发态电子轨道电子跃迁跃迁能/nm计算值实验值振子强度f计算值实验值气相S1(A2)30→31(0.70426)σ→π∗254.10.0000S2(B1)27→31(0.69474)σ→π∗213.30.0004S3(B2)29→31(0.69091)π→π∗213.2217.80.156428→31(0.48876)π→π∗S4(A1)30→32(0.49094)σ→σ∗211.40.000330→34(0.12335)σ→Ryd2溶剂(乙腈)S1(A2)29→31(0.70455)σ→π∗240.60.0000S2(B2)30→31(0.69458)π→π∗213.3217.80.19100.1303S3(B1)27→31(0.69806)σ→π∗207.00.000628→31(0.60141)π→π∗205.60.0149S4(A1)29→32(0.34502)σ→σ∗
为了阐明DMETD在气相中激发态的电子结构,8个前线分子轨道图见图3:最高占有分子轨道30(HOMO)主要两个N原子贡献形成的σ轨道,第二最高占有分子轨道29(HOMO-1)主要是由C=N双键形成的π轨道,而最低未占有空轨道31(LUMO)为π*反键轨道,而轨道27主要是N4N5的p轨道头碰头形成的σ键,轨道28是π成键轨道,轨道32是σ*,其它两个空轨道33和34都是弥散轨道(Ryd1和Ryd2),故S3的电子跃迁可归属为π →π*。在乙腈、甲醇和水溶剂中的分子轨道计算结果相同,显示S2的电子跃迁是由30(HOMO)的π轨道跃迁到31(LUMO)的π*反键轨道,如图4所示。
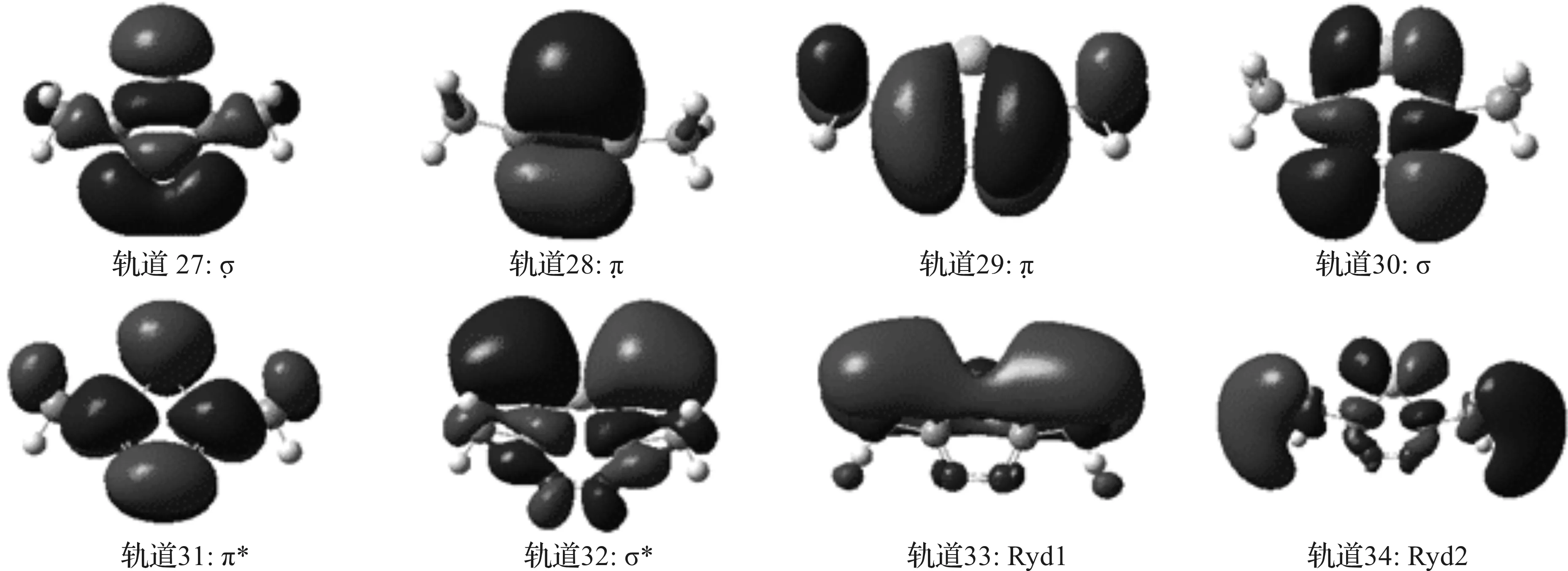
图3 DMETD 在气相中电子跃迁的分子轨道图
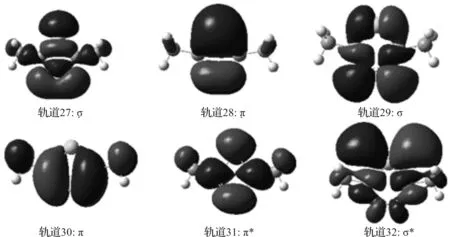
图4 DMETD在乙腈、甲醇和水溶剂中的电子跃迁的分子轨道图
3.3 共振拉曼光谱的研究
3.3.1 共振拉曼光谱的指认
目前,关于DMETD在溶剂中的共振拉曼光谱、指认和理论计算等尚无文献报道。为了探讨DMETD在不同溶剂中共振拉曼光谱的分析指认和激发态结构动力学信息,本文测定了DMETD固体FT-Raman和FT-IR的光谱图,如图5所示,相应的频率均已标在谱图上,对比发现两张图500~1500 cm-1区域内主要峰的位置吻合的较好,并结合理论计算的振动频率及拉曼活性对DMETD的振动模式进行了归属。DMETD分子是C2v对称性,有33个振动模(11个A1,6个B1,6个A2和10个B2)。表2列出了DMETD的理论计算和共振拉曼实验的振动频率,FT-Raman,FT-IR以及光谱的具体指认。
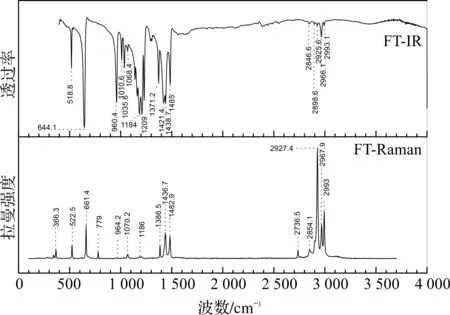
图5 DMETD固体实验的FT-IR和FT-Raman光谱图
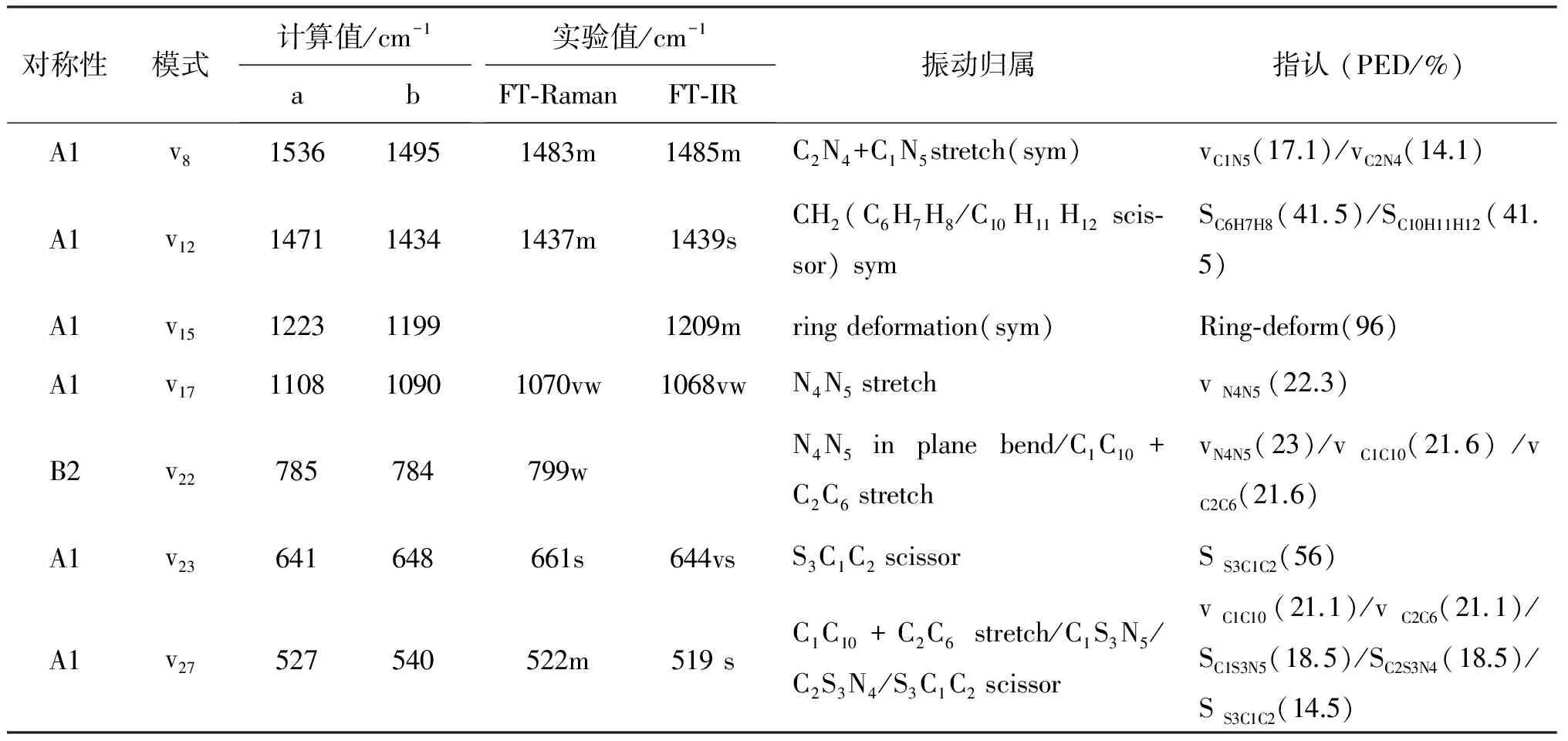
对称性模式计算值/cm-1ab实验值/cm-1FT⁃RamanFT⁃IR振动归属指认(PED/%)A1v8153614951483m1485mC2N4+C1N5stretch(sym)vC1N5(17.1)/vC2N4(14.1)A1v12147114341437m1439sCH2(C6H7H8/C10H11H12scis⁃sor)symSC6H7H8(41.5)/SC10H11H12(41.5)A1v15122311991209mringdeformation(sym)Ring⁃deform(96)A1v17110810901070vw1068vwN4N5stretchvN4N5(22.3)B2v22785784799wN4N5inplanebend/C1C10+C2C6stretchvN4N5(23)/vC1C10(21.6)/vC2C6(21.6)A1v23641648661s644vsS3C1C2scissorSS3C1C2(56)A1v27527540522m519sC1C10+C2C6stretch/C1S3N5/C2S3N4/S3C1C2scissorvC1C10(21.1)/vC2C6(21.1)/SC1S3N5(18.5)/SC2S3N4(18.5)/SS3C1C2(14.5)
注:a:Cal. = B3LYP/6-311++(d, p) calculated;b:Calc.×0.94674+41.30173;v:stretching, δ:in-plane substitute bending, γ:out-of-plane substitute bending, ρt:twist, ρs:scissor, ρw:methyl wag;sym:symmetric stretching;asym:asymmetric stretching;PED:potential energy distribution.
3.3.2 共振拉曼光谱分析
图6为DMETD在乙腈溶剂中217.8,223.1,228.7,239.5 和245.9 nm 5个激发波长下,扣除乙腈溶剂峰后的共振拉曼光谱实验对比图。图中 * 表示溶剂峰位置。从图6中可以看到不同激发波长对DMETD振动模的拉曼强度和位移影响不大。这主要是由于2,5位取代基均为CH3,与噻二唑环形成超共轭体系,增加了分子结构的稳定性(由于在甲醇和水中不同激发波长下的拉曼强度无明显变化,因此只呈现了在乙腈溶剂中的谱图)。
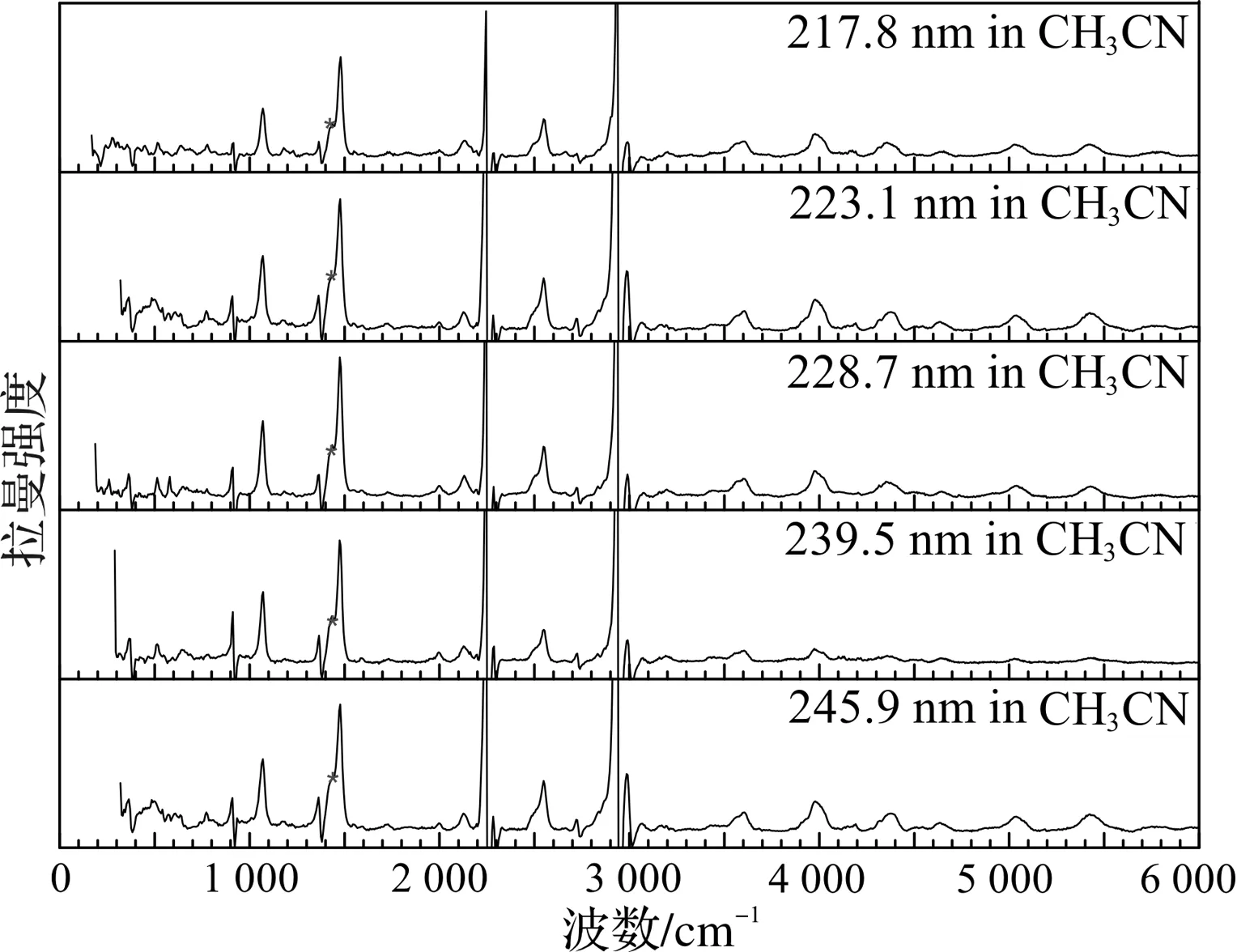
图6 DMETD在不同激发波长下乙腈溶剂中共振拉曼光谱对比图注:*为溶剂峰扣减位置。
图7为DMETD在217.8 nm激发波长下乙腈、甲醇和水溶剂中扣除溶剂峰后的共振拉曼光谱指认全图(图中*为溶剂扣减位置,#为激光线)。从图7中可以看到DMETD在三种溶剂中的拉曼强度和位移均无明显变化,故在三种溶剂中振动模式相同,跃迁主体都是πH→πL*。结合表2中振动频率的指认,DMETD在乙腈、甲醇和水溶剂中的共振拉曼光谱大致可以被指认为C2N4和C1N5的对称伸缩振动(v8),CH2对称(C6H7H8/C10H11H12剪式振动) (v12), 环变形(v15),N4N5伸缩振动(v17),N4N5面内摆动和C1C10+C2C6伸缩振动(v22),S3C1C2剪式振动(v23)和C1C10+C2C6伸缩/ C1S3N5/C2S3N4/S3C1C2剪式运动(v27)这7个Frank-Condon区域的活动振动模的基频、倍频和它们的组合频。然而C=N和N-N伸缩振动的基频、倍频和组合频占据了吸收带的共振拉曼的主要部分,说明在不同的溶剂中A吸收带的激发态结构动力学主要沿这两个振动模的初始反应坐标进行。其S2激发态的衰变动力学可能是S2,FC→S2/S1→S0或S2,min→S2/S1→S0光物理过程,这有待于进一步的理论计算。
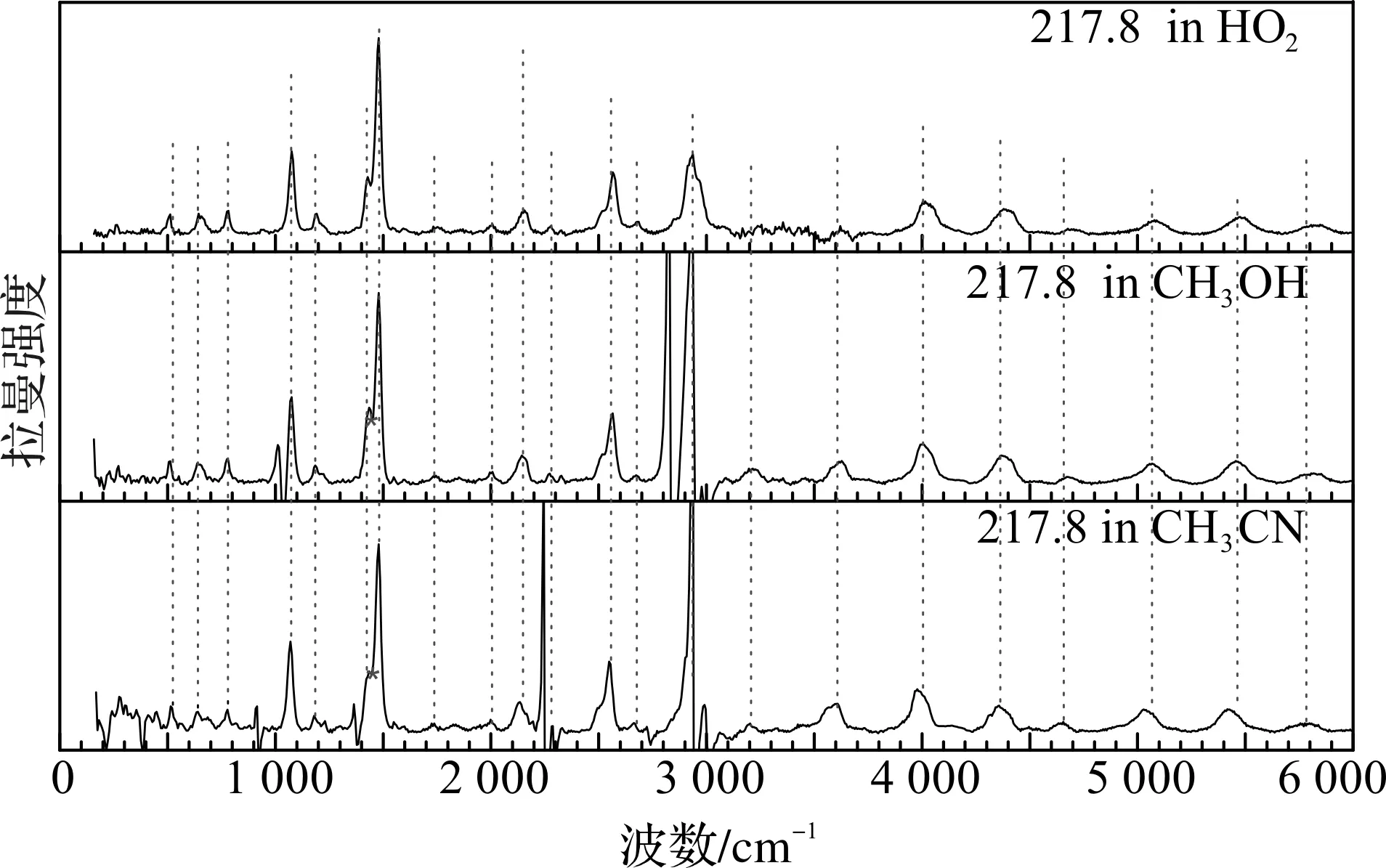
图7 DMETD在217.8 nm激发波长下乙腈、甲醇和水溶剂中的共振拉曼光谱指认全图
4 结 论
本文采用共振拉曼光谱技术联合密度泛函理论计算对2,5-二甲基-1,3,4-噻二唑(DMETD)的激发态结构动力学进行了研究,得出如下结论:
a)DMETD由于两个甲基与噻二唑环形成超共轭,使其结构非常稳定,因此在溶剂中的构型与溶剂极性无关。
b)DMETD在气相和溶剂中的跃迁主体都是π→π*,溶剂对该主体跃迁无影响。
c)DMETD在乙腈、甲醇和水中的共振拉曼光谱图大致可以指认为7个Frank-Condon区域的活动振动模的基频、倍频以及它们的组合频和泛频,然而C=N和N-N伸缩振动的基频、倍频和组合频占据了共振拉曼吸收带的主要部分,故S2激发态结构动力学是沿着这两个振动模的反应坐标进行的。
[1] BALABAN A T, ONICIU D C, KATRITZKY A R. Aromaticity as a cornerstone of heterocyclic chemistry[J]. Chemical Reviews,2004,104(5):2777-2812.
[2] JAIN S K, MISHRA P. Original research article appraisal of analgesic and anti-inflammatory activity of some 2, 5-disubstituted-1, 3, 4-thiadiazoles[J]. Int. J. Curr. Microbiol. App. Sci,2014,3(10):849-855.
[3] BANDAY M R, RAUF A. Synthesis and evaluation of in vitro antibacterial activity of novel 2, 5-disubstituted-1, 3, 4-thiadiazoles from fatty acids[J]. Chinese Chemical Letters,2008,19(12):1427-1430.
[4] YUSUF M, KHAN R A, AHMED B. Syntheses and anti-depressant activity of 5-amino-1, 3, 4-thiadiazole-2-thiol imines and thiobenzyl derivatives[J]. Bioorganic & Medicinal Chemistry,2008,16(17):8029-8034.
[5] WEI M X, FENG L, LI X Q, et al. Synthesis of new chiral 2, 5-disubstituted 1, 3, 4-thiadiazoles possessing γ-butenolide moiety and preliminary evaluation of in vitro anticancer activity[J]. European Journal of Medicinal Chemistry,2009,44(8):3340-3344.
[6] ORUC E E, ROLLAS S, KANDEMIRLI F, et al. 1, 3, 4-Thiadiazole derivatives. Synthesis, structure elucidation, and structure-antituberculosis activity relationship investigation[J]. Journal of Medicinal Chemistry,2004,47(27):6760-6767.
[7] KHAN I, ALI S, HAMEED S, et al. Synthesis, antioxidant activities and urease inhibition of some new 1, 2, 4-triazole and 1, 3, 4-thiadiazole derivatives[J]. European Journal of Medicinal Chemistry,2010,45(11):5200-5207.
[8] 刘琳,潘晓娜,张强,等.噻二唑衍生物分子结构与其缓蚀性能的关系[J].化工学报,2014,65(10):4039-4048.
[9] HUANG L, TANG F, HU B, et al. Chemical reactions of 2, 5-dimercapto-1, 3, 4-thiadiazole (DMTD) with metallic copper, silver, and mercury[J]. The Journal of Physical Chemistry B,2001,105(33):7984-7989.
[10] ROSTKOESKA H, LAPINSKI L, NOWAK M J. UV-induced transformations of matrix-isolated 1,3,4-thiadiazole-2-thiones[J]. Journal of Physical Organic Chemistry,2010,23(1):56-66.
[11] 武海顺,许小红,马文瑾,等.AMT异构体互变机理的理论研究[J].物理化学学报,2003,19(5):408-413.
[12] ZHENG X M, PHILLIPS D L. Effect of geometrical conformation on the short-time photodissociation dynamics of 1-iodopropane in the A-band absorption[J]. The Journal of Chemical Physics,1998,108(14):5772-5783.
[13] ZHENG X M, LI Y L, PHILLIPS D L. Resonance Raman study of the short-time photodissociation dynamics of the a-band absorption of cyclopropyl iodide in cyclohexane solution[J]. The Journal of Physical Chemistry A,2004,108(39):8032-8039.
[14] ZHU X M, ZHANG S Q, ZHENG X, et al. Resonance Raman study of short-time photodissociation dynamics of the charge-transfer band absorption of nitrobenzene in cyclohexane solution[J]. The Journal of Physical Chemistry A,2005,109(13):3086-3093.
[15] JIAN J W, ZHANG H B, CHEN C Q, et al. Resonance Raman spectroscopic and density functional theory investigation of the excited state structural dynamics of 2-mercapto-1-methylimidazole[J]. Journal of Raman Spectroscopy,2013,44(4):582-589.
[16] MYERS A B, LI B, CI X. A resonance Raman intensity study of electronic spectral broadening mechanisms in CS2/cyclohexane[J]. The Journal of Chemical Physics,1988,89(4):1876-1886.
[17] FRISCH M, TRUCKS G W, SCHLEGEL H B, et al. Gaussian 09, Revision A. 02, Gaussian[J]. Inc., Wallingford, CT, 2009, 19:227-238.
(责任编辑: 许惠儿)
Resonance Raman Spectroscopic Investigation of Excited State Structural Dynamics of 2, 5-dimethyl-1, 3, 4-thiadiazole
PANGSumei,ZHAOYanying
(School of Sciences, Zhejiang Sci-Tech University, Hangzhou 310018, China)
The ultraviolet absorption spectra and resonance Raman spectra of 2, 5-dimethyl-1,3,4-thiadiazole (DMETD) were obtained in acetonitrile, methanol and water. Short-time structural dynamics of DMETD was studied in combination of density functional theory TD-B3LYP/6-311++G(d,p). Resonance Raman spectra of DMETD in the solvents were identified through solid-state FT-Raman, FT-IR spectrum and DFT calculation. Besides, the intensity mode of Resonance Raman spectra of DMETD was analyzed. The results show that transition subject of DMETD in gas phase and solvents isχ→χ*. Solvent model calculation indicates that the polarity of solvents has no influence on Raman intensity. Thus, the active vibrational mode of DMETD in the Franck-Condon region can be identified as 7 fundamental frequencies (v8, v12, v15, v17, v22, v23and v27), their frequency doubling and combined frequencies. C=N and N-N stretching vibrations occupy the main part of Raman intensity. This indicates that S2excited state structural dynamics is mainly along the two coordinates.
2, 5-dimethyl-1, 3, 4-thiadiazole; UV spectra; resonance Raman spectra; density functional theory; excited state structural dynamics
10.3969/j.issn.1673-3851.2017.01.021
2015-12-09
日期: 2016-12-09
国家自然科学基金项目(21273202,21473162)
庞素梅(1990-),女,山西晋中人,硕士研究生,主要从事化学反应动力学方面的研究。
赵彦英,E-mail: yyzhao@zstu.edu.cn
O643.12
A
1673- 3851 (2017) 01- 0127- 06
猜你喜欢
辽宁省博物馆馆刊(2021年0期)2021-07-23
汕头大学学报(自然科学版)(2020年4期)2020-12-14
江苏理工学院学报(2020年2期)2020-10-23
物理学报(2020年16期)2020-08-29
科技视界(2019年27期)2019-11-05
食品界(2017年3期)2017-04-19
新疆人文地理(2016年9期)2016-05-14
国外科技新书评介(2014年8期)2014-12-05
读写算·教研版(2014年12期)2014-09-01
