
线粒体基因组变异与衰老和疾病
2017-01-09 08:54梁庆华胡才友何本进潘尚领
中国老年保健医学 2016年6期
梁庆华 胡才友 何本进 潘尚领 杨 泽
线粒体基因组变异与衰老和疾病
梁庆华1胡才友1何本进1潘尚领2杨 泽3※
线粒体基因组的遗传特点呈母系传递。线粒体有自己独立的基因组,并且线粒体DNA(mtDNA)具有与核基因组不同的结构和代码,mtDNA基因组编码可产生部分线粒体呼吸链的多肽蛋白。如果mtDNA发生突变,依照其表型受累的程度,可将其归类为:①严重程度,为罕见、高遗传(保守)性的因果性突变,常引起一系列神经系统、肌肉、心脏和内分泌器官严重的人类单基因疾病;②中等程度或轻度突变,常引起常见的复杂性疾病和迟发性及老年相关疾病,如帕金森氏病和阿尔兹海默病。另外,多数健康人也会带有低水平(<1%)的mtDNA点突变,包括先天遗传性突变和后天获得性突变。其中随着获得性突变数量的增加,当超过特定阈值水平时,会导致老年相关疾病的发生。本文内也依据现有模式生物的研究结果,提供有人们对mtDNA突变机制的新知识。
线粒体基因组 变异 衰老 疾病
大约25年前,人们发现可遗传的线粒体DNA(mtDNA)突变是人类疾病的主要原因。最初认为mtDNA突变是极为罕见的,现在已知mtDNA疾病患病率在人群中为1/4300,并且已报道有数百种不同的点突变,mtDNA缺失与广泛的、重叠的临床表型相关联。mtDNA疾病的发生范围从新生儿到老年人,并且经常影响神经系统。同时,mtDNA的体细胞突变有随着年龄累积的特点,尤其多见于几种常见晚发疾病的受累器官之中。mtDNA的体细胞突变在神经退行性疾病中的作用特别引人注目,最近的支持性证据表明,遗传性和获得性mtDNA突变可以相互作用并引起衰老表型。而且,独立的遗传关联研究表明,几种晚发的退行性疾病与常见遗传多态性(mtDNA12)关联。虽然这些多态性被认为没有任何表型效应,但新的证据表明,他们可以改变线粒体的功能,影响了器官依赖的线粒体能量代谢,从而改变了发生常见疾病的风险。尚不清楚如何将这些不同的观察结果统一起来,但最近的观察已经揭示了它们之间具有相互连接的机制。mtDNA的常见遗传变异增加了一些重大健康问题发生的风险,如神经退行性疾病、帕金森氏病和阿尔兹海默病。本文中,我们解释了mtDNA变异如何发生,以及单个细胞中的单个分子来源的mtDNA变异如何扩散到整个特定器官中并达到高水平存在,且最终促发了常见的老年相关疾病。尽管mtDNA有高突变率,并且复制增殖几乎没有分子重组,但是mtDNA始终存在于所有需氧真核生物中。尽管最近的研究发现,在雌性生殖细胞中mtDNA新突变受到抑制;但是,mtDNA异质性的存在是非常普遍的。哺乳动物mtDNA的遗传瓶颈结果显示,生殖细胞mtDNA新突变可以在一代内增加到高水平,并通过固定那些不太严重的突变在mtDNA内,逃避了生殖细胞选择,贡献着老年相关疾病的发病风险。
在本文中,我们描述了从线粒体内单分子的起源,到人群mtDNA变异,均有共同的mtDNA突变的特征,并且说明了mtDNA突变的机制,及其遗传和后续固定在母系胚系中的机制,这些知识对我们理解人类疾病和衰老是重要的。这些发现质疑着我们对细胞核和线粒体之间复杂关系的认识,并提出了与人类进化相关的问题。
1.线粒体的生物学发生和mtDNA
大约20亿年前,一个原始的蛋白质细菌与一个原核细胞相融合,产生了一种生物学中最持久的共生关系。在被细胞内吞后,蛋白质细菌成为双膜的细胞质细胞器,并逐渐将遗传物质扩散传递到原核细胞核中。数百万年来,这个过程减少了细胞器内原始线粒体基因组数量的整体大小,巩固了细胞器(后来成为线粒体)与所有真核细胞核之间的共生关系。
线粒体在细胞能量代谢中发挥中心作用,在细胞内钙信号传导和铁硫簇的生成中是重要的,并且参与细胞凋亡。人线粒体保留了16.5kb的双链DNA-mtDNA分子,编码了氧化磷酸化系统的13个核心肽亚基和线粒体内蛋白质合成所必需的24个RNA(图1)。然而,大多数线粒体蛋白(总数估计约1500个)由位于细胞核中的基因转录,在胞浆中翻译后进入线粒体,线粒体内有低于10%的细胞蛋白质组。由于人类线粒体的最佳功能至关重要地依赖于这两个不同的遗传系统,因此自然选择引起的线粒体基因或核基因中的突变均可以增强或减弱线粒体功能。
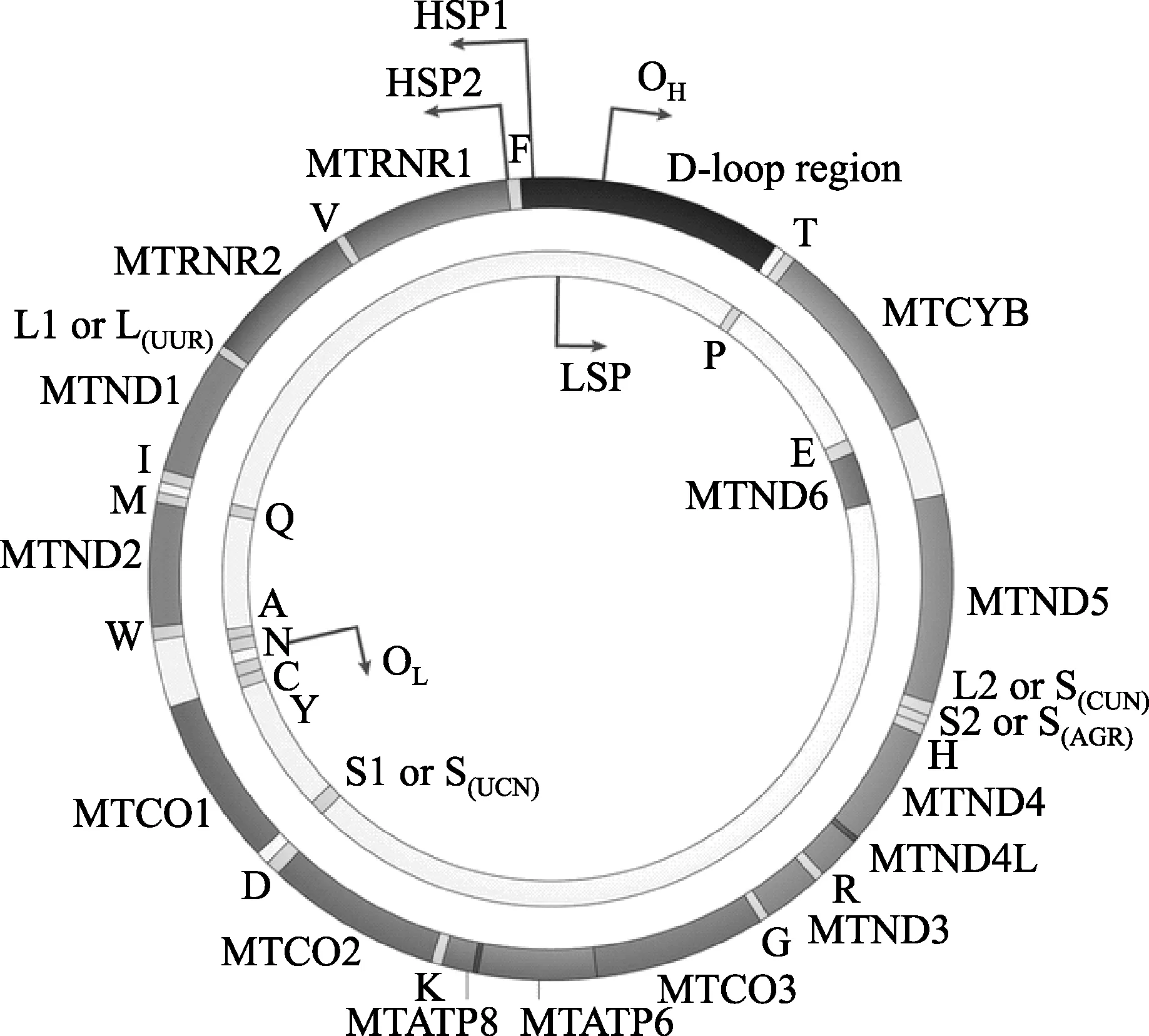
图1 人类mtDNA的基因结构和编码蛋白质
2.人类mtDNA的基因结构和编码蛋白质
人类mtDNA基因组包含16,569个碱基对,组成了内部的“轻”(L)链和外部的“重”(H)链。轻、重链代表了影响分子量的鸟嘌呤含量,见图1。编码线粒体呼吸链结构亚基的mtDNA基因包括:线粒体编码的NADH脱氢酶1(MTND1)-MTND6和MTND4L(复合体I);细胞色素b(MTCYB;复合体III);细胞色素c氧化酶I(MTCO1)-MTCO3(复合体IV);和ATP合酶6(MTATP6)和MTATP8(复合体V)。另有22个tRNA和2个rRNA基因散布在蛋白编码基因之间。
mtDNA进行复制时,通过多蛋白复制体,包括mtDNA聚合酶-γ(Polγ),解旋酶(Twinkle;也称为PEO1),拓扑异构酶I,mtDNA单链DNA结合蛋白等。目前有两种mtDNA复制模型,分别为“链置换”和“不同步”模型。链置换复制是通过非编码mtDNA内的置换(displacement,D)环区启动转录复制,并从重链复制起始点(OH;也称为OriH)沿顺时针方向进行,一直复制到轻链(OL)起始点出现,轻链合成接着沿顺时针方向进行,直到整个mtDNA分子被复制完成。不同步复制是指对称链偶联复制,在某些情况下也可能发生;这时,复制起始于分布在D环区4kb片段3′端的多个起点上,并在复制“环”中沿两个方向进行复制。复制在OH处停止,并允许在一个方向上复制分子的其余部分。
人类mtDNA有三个转录启动子:重链启动子1(HSP1)启用两个核糖体RNA的转录;HSP2启动其余重链的转录作为大的多顺反子转录物,然后被剪接成功能性tRNA,rRNA和mRNA;LSP通过RNA酶加工线粒体RNA(MRP)通过启动轻链转录为一个长转录物或几个小引物的转录物。这些引物连接mtDNA转录复制。启动转录涉及几种蛋白质,包括线粒体RNA聚合酶和线粒体转录因子A和B2。
从原始共生事件发生以来,mtDNA和核DNA之间出现了几个差异。mtDNA遗传密码与核基因组的遗传密码不同。对于脊椎动物mtDNA,密码子AUA和AUG都编码甲硫氨酸,UGA编码色氨酸(不是核基因组中的终止密码子),并且读取AGA和AGG作为终止密码子(在核基因组中是精氨酸)。此外,mtDNA通常保持环状和紧密的编码排列,代替核内成对的染色体。在人类每个细胞内存在有许多独立增殖的mtDNA拷贝,mtDNA拷贝数范围从精子中的低于100个拷贝到未受精的卵母细胞中的数十万个。拷贝数受到严格调控,并且在一些细胞类型中可随时间而发生改变(如:肌肉细胞中mtDNA拷贝的数量可随运动而增加)。蛋白质包裹着mtDNA一起入核,每个核中均包含一或两个mtDNA分子。最后,尽管在线粒体内存有修复元件,但是一般认为mtDNA修复不如核DNA修复有效,但细胞功能仍可正常,部分是因为高拷贝数缓冲了随机突变事件的有害影响。
3. mtDNA异质性和阈值效应
假定每个细胞内有多拷贝的mtDNA,如突变影响到所有分子,称为同质性;如仅一定比例的分子,称为异质性(图2)。异质性水平是指在同一组织或器官内的不同细胞之间,同一个体内不同器官之间,及同一家族不同个体之间的mtDNA变异水平的不同。由于mtDNA仅沿母系遗传,故可忽略其在群体水平表现的分子间重组,因此mtDNA谱系扩展在很大程度上被认为是复制。在大约三代人中发生的线粒体DNA(mtDNA)突变通常是异质性的,并且同一个细胞内也可以包含有不同比例的突变型和野生型mtDNA。如果是致病性突变,则在突变水平超过生化阈值水平并且检测到有呼吸链缺陷之前,细胞通常可以耐受一定比例的致病性突变。一般来说,突变阈值水平>80%,则表明这些mtDNA突变,大多是单倍剂量不足或隐性突变。
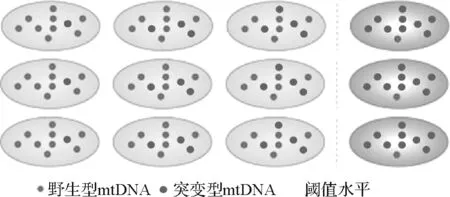
图2 mtDNA异质性和阈值效应
4.人类单基因疾病
mtDNA突变与人类疾病的第一个明确致病突变的证据,来自20世纪80年代后期对不明原因的神经和神经肌肉疾病患者的研究,发现编码呼吸链复合体I,线粒体编码的NADH脱氢酶4的亚基1的MTND4,m.11778G>A的同义点突变,在Leber遗传性视神经病(LHON)的母系大家系中有遗传易感性倾向,并在生物化学定义的线粒体肌病个别病例中,发现患者携有mtDNA2缺失的大的异质性。
随着分子技术的进步,接下来的几十年识别了多种临床疾病表型患者中的几百个mtDNA的点突变和缺失(图3)。尽管有临床表型异质性和遗传异质性,但是已经发现了mtDNA突变的几种趋势:①mtDNA点突变:通常为母系遗传,同一家族中有多个成员受累。②mtDNA片段缺失:很少遗传(可能只通过反复中间重排),且缺失没有同质性。③mtDNA同质点突变:通常引起相对温和的生物化学缺陷,且仅影响一个器官或组织(如:LHON,耳聋或心肌病)。④异质突变:影响多器官系统,特别是脑、脊髓、肌肉、外周神经、心脏和内分泌器官。异质性水平与器官受累的程度相关,与临床表型的严重程度相关,因为通常受累组织中有严重的生化缺陷。
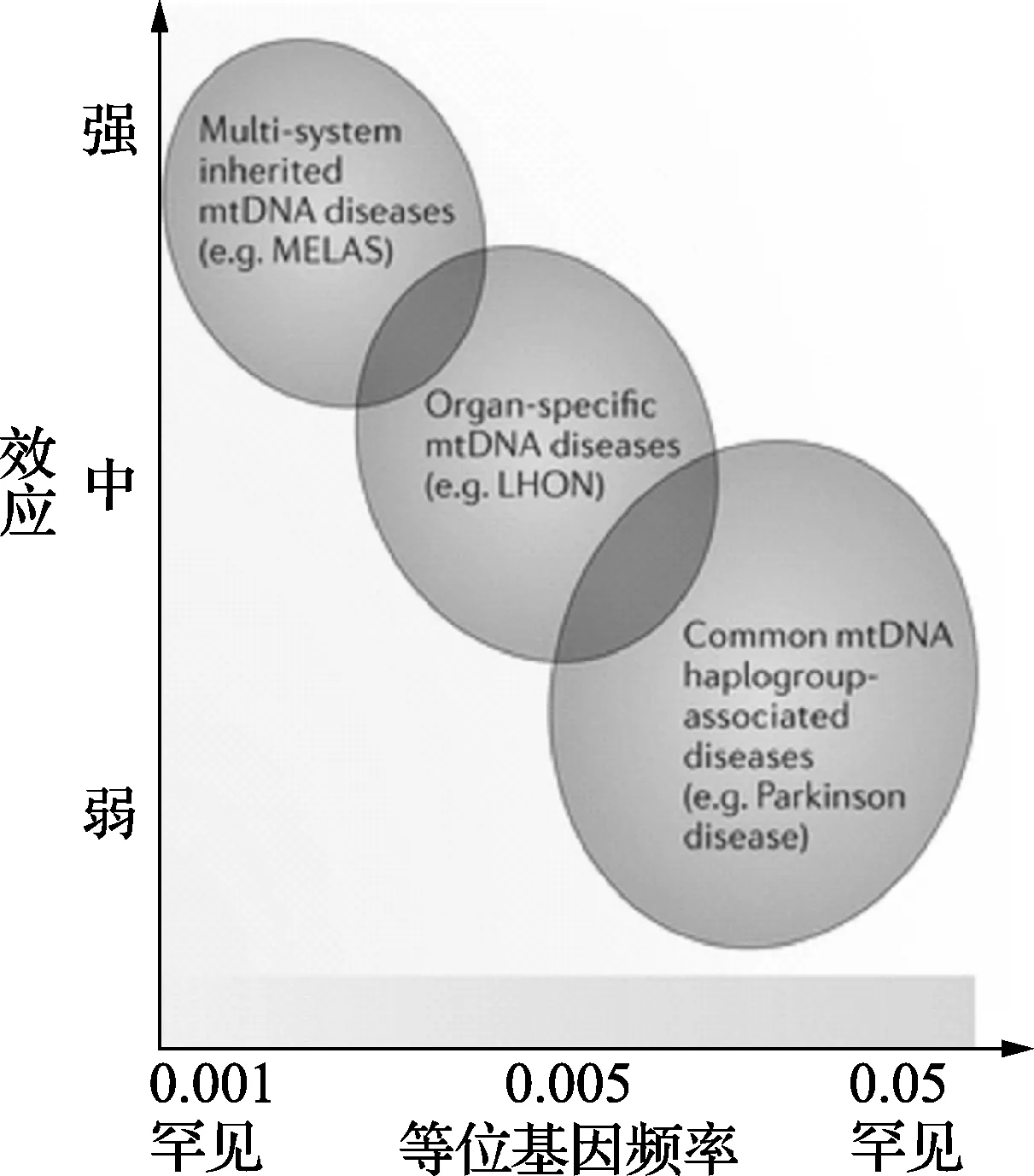
图3 mtDNA变异范围及其对人类疾病的影响
线粒体DNA(mtDNA)变异如果在连续范围内,从单基因疾病的因果变异,到常见复杂疾病的风险等位基因,再伴有上位效应和环境因素一起相互作用,会极大地影响着临床外显率的变化(见图3)。最令人信服的证据来自帕金森氏病研究,对于一般人群中的多基因疾病,常见变异对发生常见疾病的风险影响不大。几项mtDNA的常见变异影响帕金森氏病发病机制的大型研究结果提示着同一个mtDNA变异,可能共有不同疾病,并有同一个线粒体风险机制。共有变异通常是古老的和保守的变异(>10,000岁),因此是同质变异。还有一些变异位于罕见和常见变异两个极端之间,并且仅当与其他遗传因素或环境因素共存时,联合作用才会导致疾病,如:m.11778A>G,m.3460A>G和m.14484T>CmtDNA突变联合作用引起Leber遗传性视神经病(LHON),而mtDNA单倍型组,核基因组和环境因素(如吸烟)等因素影响着LHON的临床外显率。通常这些是同质突变,但不是单一的突变。非常罕见的高外显率mtDNA突变,是多系统线粒体遗传病的主要原因,包括由m.3243A>G突变引起的乳酸酸中毒和m.8344A>G突变引起的线粒体脑肌病(MELAS);由4,977bpmtDNA的大片段缺失引起的Kearns-Sayre综合征。点突变通常会影响两代或三代人,但mtDNA异质性片段缺失,一般仅影响一代人。
对于异质突变,体外研究使用杂交细胞系和使用来自组织活检样品分离的单细胞进行研究,已经显示mtDNA的突变比例必须超过临界阈值水平(通常为60%~80%),然后可以使用已建立的实验室技术检测线粒体的生化缺陷。不同的突变及同一突变在不同组织中的阈值水平不同。异质性水平和精确阈值的差别被认为有助于解释在不同线粒体疾病中器官易感性的特征模式和在携带相同mtDNA突变的不同患者中观察到的临床异质性。
虽然一些新的证据表明环境和上位因素可能影响线粒体疾病的临床表现,特别是mtDNA同质突变,是表型所必需的主要mtDNA突变,所以线粒体疾病也被认为是单基因疾病(图3)。流行病学研究表明,这些原发性mtDNA疾病影响约1/4300人,其中最常见的是遗传代谢性疾病。但难以说明的是,随后发现10个最常见的致病性mtDNA点突变携带率,在健康个体中约为1/200,且异质性水平低。研究者使用的技术可以可靠地检测特异性mtDNA等位基因和异质性水平>1%。然而,这些观察给出了潜在致病性mtDNA突变在人群中的真实频率,并且提出了这些突变会如何发生?及如何从单一分子最终增加到引起已知疾病的高百分比突变的异质性阈值水平发生的问题。
5.mtDNA的体细胞突变和疾病
在原发性mtDNA疾病患者中,所有突变分子具有完全相同的突变。然而,不论是从老年人还是从健康人取得的活检组织均有极低水平的几种不同mtDNA突变,并且在同一健康老年人的有丝分裂后(非分裂)组织中检测到了mtDNA缺失和mtDNA的点突变,虽然组织或器官的总体突变率低,但个体突变可在单个细胞内累积至更高水平,从而引起生物化学缺陷。之后随时间发展,生物化学缺陷细胞的比例数量增加,这些缺陷增加可能促进了老化的过程。
6.发生机制
在几种迟发性退行性变疾病(如帕金森病和阿尔茨海默病)患者的器官中观察到高频率的生物化学缺陷细胞,与这些常见人类神经退行性疾病发病机制中的体细胞突变有关联。这些突变参与的发病机制得到了动物模型数据的支持,其中过多的遗传性突变或新突变的比例数量可导致典型的老年相关疾病。
在癌症患者中也观察到mtDNA突变,提示着mtDNA突变可能在肿瘤发生或转移扩散中起作用。这一观点是有吸引力的,因为已知恶性肿瘤中的氧化代谢转变为无氧糖酵解(称为Warburg效应),并且得到不同类型癌症中特异性mtDNA突变证据的支持。然而,迄今为止最大规模研究人类癌症mtDNA突变的证据并不支持这一观点。在一项1675个肿瘤组织中观察到mtDNA突变的不寻常模式,多数突变聚集在mtDNA复制的前导链,类似于在人群中观察到的mtDNA同质突变模式。这表明mtDNA突变既不会引起恶性肿瘤,也不贡献恶性肿瘤转移的过程,而仅是反映了在快速增殖的恶性细胞内mtDNA复制期间错误分子的堆积。
最近,下一代测序(NGS)方法已被用于同时识别和定量mtDNA突变(图2)。然而,在用于检测和测量低水平异质性所需要的极高深度时,即使短读长测序技术也有高的内在错误率,长读长技术的内在错误率则更高。不论使用短读长或是长读长测序技术,测序数据均可能被核内的线粒体DNA序列(NUMT)所污染或测序产生的DNA碱基损伤,可能被误识别为“真实的”序列变异。NUMTs的个体水平变化的证据表明,当使用下一代短读长测序数据时,在分析过程中可能极难过滤掉多态的NUMT。但是可以使用长扩增子方法和捕获-富集方法,虽然不能完全消除数据中的NUMT。应用密度梯度离心法富集mtDNA会带来高水平的核内DNA的污染。尽管使用这些技术方法有缺陷,但在几乎每一个已经研究的健康个体中都发现有mtDNA的异质性,虽然水平非常低。尽管,NGS提高了在生命晚期检测体细胞组织(或肿瘤)中体细胞突变的可能性,实际上检测到的这些突变是经母系遗传的低水平异质性变异。但是,小鼠模型的研究已经表明,线粒体低水平异质性变异可以与体细胞新突变相互作用,加剧衰老和神经退行性疾病的发生。
1 Wallace DC, Singh G, Lott MT, et al. Mitochondrial DNA mutation associated with Leber’s hereditary optic neuropathy[J]. Science, 1988, 242(4884): 1427-1430. DOI:10.1126/science.3201231.
2 Holt IJ, Harding AE, Morgan-Hughes JA. Deletions of muscle mitochondrial DNA in patients with mitochondrial myopathies[J]. Nature, 1988, 331(6158): 717-719.
3 Gorman GS, Schaefer AM, Ng Y, et al. Prevalence of nuclear and mitochondrial DNA mutations related to adult mitochondrial disease[J]. Ann Neurol, 2015, 77(5): 753-759.
4 Chinnery PF, Johnson MA, Wardell TM, et al. The epidemiology of pathogenic mitochondrial DNA mutations[J]. Ann Neurol, 2000, 48(2): 188-193. DOI:10.1002/1531-8249(200008)48:2<188::aid-ana8>3.3.co;2-g.
5 DiMauro S, Schon EA, Carelli V, et al. The clinical maze of mitochondrial neurology[J]. Nat Rev Neurol, 2013, 9(8): 429-444.
6 Vafai SB, Mootha VK. Mitochondrial disorders as windows into an ancient organelle[J]. Nature, 2012, 491(7424): 374-383.
7 Wallace DC. Colloquium paper: bioenergetics, the origins of complexity, and the ascent of man[J]. Proc Natl Acad Sci USA, 2010, 107(Suppl 2): 8947-8953.
8 Pinto M, Moraes CT. Mechanisms linking mtDNA damage and aging[J]. Free Radic Biol Med, 2015, 85: 250-258.
9 Pinto M, Moraes CT. Mitochondrial genome changes and neurodegenerative diseases[J]. Biochim Biophys Acta, 2014, 1842(8): 1198-1207.
10 Ross JM, Stewart JB, Hagström E, et al. Germline mitochondrial DNA mutations aggravate ageing and can impair brain development[J]. Nature, 2013, 501(7467): 412-415.
11 Keogh M, Chinnery PF. Hereditary mtDNA heteroplasmy: a baseline for aging[J]. Cell Metab, 2013, 18(4): 463-464.
12 Hudson G, Gomez-Duran A, Wilson IJ, et al. Recent mitochondrial DNA mutations increase the risk of developing common late-onset human diseases[J]. PLoS Genet, 2014, 10(5): e1004369.
13 Wallace D C, Chalkia D. Mitochondrial DNA Genetics and the Heteroplasmy Conundrum in Evolution and Disease[J]. Cold Spring Harbor Perspectives in Biology, 2013, 5(11): a021220-a021220.
14 Gómez-Durán A, Pacheu-Grau D, López-Gallardo E, et al. Unmasking the causes of multifactorial disorders: OXPHOS differences between mitochondrial haplogroups[J]. Hum Mol Genet, 2010, 19(17): 3343-3353.
15 Pagliarini DJ, Calvo SE, Chang B, et al. A mitochondrial protein compendium elucidates complex I disease biology[J]. Cell, 2008, 134(1): 112-123.
16 Kukat C, Wurm CA, Spåhr H, et al. Super-resolution microscopy reveals that mammalian mitochondrial nucleoids have a uniform size and frequently contain a single copy of mtDNA[J]. Proc Natl Acad Sci USA, 2011, 108(33): 13534-13539.
17 Vafai SB, Mootha VK. Medicine. A common pathway for a rare disease[J]. Science, 2013, 342(6165): 1453-1454.
18 Anderson S, Bankier AT, Barrell BG, et al. Sequence and organization of the human mitochondrial genome[J]. Nature, 1981, 290(5806): 457-465. DOI:10.1038/290457a0.
19 Andrews RM, Kubacka I, Chinnery PF, et al. Reanalysis and revision of the Cambridge reference sequence for human mitochondrial DNA[J]. Nat Genet, 1999, 23(2): 147.
20 Calvo SE, Mootha VK. The mitochondrial proteome and human disease[J]. Annu Rev Genomics Hum Genet, 2010, 11: 25-44.
Aging,Diseases and Mitochondrial Genome Variation
(LIANG Qinghua1, HU Caiyou1, HE Benjin1, PAN Shangling2, YANG Ze3
.1.Jiangbin Hospital of Guangxi Zhuang Autonomous Region, Nanning 530021, China; 2.Department of Pathophysiology, Guangxi Medical University, Nanning 530021, China; 3.Key Laboratory of Geriatrics, Chinese Ministry of Health, Beijing Hospital, National Gerontology Center, Beijing 100730, China.)
The genetic characteristics of mitochondrial genome are maternal transmission.Mitochondria have their own independent genome,and mitochondrial DNA (mtDNA) has a different structure and codes from the nuclear genome.The mtDNA encodes a polypeptide protein that produces a part of components in mitochondrial respiratory chain.If mtDNA is mutated,it is classified as:①severity,a rare,high genetic (conserved) causal mutation,which causes a series of neurological,muscular,cardiac,and endocrine disorders,depending on the extent of phenotypic involved organism serious human monogenic disease;②moderate or mild mutation,often caused by the common complex diseases and late and old age-related diseases.In addition,most healthy individuals also carry low levels (<1%) of mtDNA point mutations,including congenital and acquired mutations.With the increase in the number of acquired mutations,when a certain threshold level is exceeded,a late onset of disease may occur.In this paper,we also provide some new knowledge about mtDNA mutation mechanism based on the results of existing model organisms.
mitochondrial genome, mutation, senescence, disease
梁庆华,男,学士学位,副主任技师,主要研究领域为基因多态性及相关生物学标志物与老年痴呆的关联研究。
1.广西壮族自治区江滨医院 530021 2.广西医科大学 病理生理学教研室 530021 3.北京医院,国家老年医学中心,卫生部老年医学重点实验室 100730
国家高技术研究发展计划(863计划)项目子课题基金(基金编号2014AA022304),国家自然科学基金(81061120527,81370445,81472408),国家科技部十二五支撑计划项目(2012BAI10B01, 2015BAI06B03)。
10.3969/j.issn.1672-4860.2016.06.002
2016-10-30
※为通讯作者
猜你喜欢
临床肝胆病杂志(2022年8期)2022-11-23
中华实用诊断与治疗杂志(2022年1期)2022-08-31
中国临床医学影像杂志(2022年6期)2022-07-26
海洋通报(2021年1期)2021-07-23
现代企业(2021年2期)2021-07-20
生物学通报(2021年4期)2021-03-16
趣味(数学)(2020年4期)2020-07-27
支部建设(2020年15期)2020-07-08
铁道学报(2018年5期)2018-06-21
百科知识(2015年18期)2015-09-10
