
第三代测序技术的主要特点及其在病毒基因组研究中的应用
2017-01-05 11:00:26袁易王铭杰张欣欣
微生物与感染 2016年6期
袁易,王铭杰,张欣欣
上海交通大学医学院附属瑞金医院临床病毒研究室,上海 200025
·综述·
第三代测序技术的主要特点及其在病毒基因组研究中的应用
袁易,王铭杰,张欣欣
上海交通大学医学院附属瑞金医院临床病毒研究室,上海 200025
随着基因测序技术的创新和应用,新的高通量测序技术不断涌现,以Pacific Biosciences(PacBio)公司的单分子实时测序(single molecule real time sequencing)为代表的第三代测序(third generation sequencing,TGS)技术开始逐渐应用于基因组研究,包括大型基因组拼装、基因结构变异和表观遗传研究等方面。本文主要对TGS技术的原理、特点和应用,特别是在病毒研究中的应用进行介绍,并与第二代测序(next generation sequencing,NGS)技术进行比较,为基因组测序技术的选择及其临床应用提供一定参考。
第三代测序技术;Pacific Biosciences;单分子实时测序;病毒基因组
近几年来基因测序技术发生了重大革新,出现了以高通量、相对较低成本为特征的第二代测序(next generation sequencing,NGS)技术,并广泛应用于生物医学领域,如基因组重测序(genome resequencing)、从头测序(denovoassembling)、转录组测序(transcriptome sequencing)、外显子测序(exome sequencing)等方面[1]。但NGS技术在实际应用中也存在缺陷,如读长较短,无法跨越基因上串联重复区域,导致其在检测串联重复区域时存在明显不足[2]。与NGS技术相比,Pacific Biosciences(PacBio)公司推出的最新单分子实时测序(single molecule real time sequencing,SMRT)具有读长较长的优势,平均读长10~15 kb,最大读长达64.5 kb[3],属第三代测序(third generation sequencing,TGS)技术。目前,病毒基因组的测序工作主要采用NGS技术,最常用的是Roche454测序平台和Illumina测序平台。这两种测序平台存在的主要问题是读长较短,其中读长最长的 Roche454 GS FLX+平台的读长也仅约700 bp[4]。因此,对读长要求较高的特殊分析如检测联合变异时灵敏度较低。TGS技术的读长优势可弥补NGS技术的不足,可用于病毒基因组研究,加深人类对病毒变异的了解,对病毒感染性疾病的治疗和预后具有重要意义。
1 PacBio SMRT测序
1.1 PacBio测序原理
PacBio测序平台采用的检测单位为SMRT cell,每个SMRT cell中含有15万个零模式波导管(zero-mode waveguide,ZMW)。ZMW是一种孔径只有100 nm的小孔,内径比激发光波长小得多。从ZMW底部射出的激发光无法穿过小孔进入上方的溶液中激发游离碱基产生荧光,可最大程度地降低背景荧光的干扰。PacBio测序的文库是由双链DNA(double-strand DNA,dsDNA)分子在两端分别结合标记性的接头后形成的发夹状产物。
测序基本原理如图1所示,构建的SMRT cell扩散至ZMW底部后双链打开,形成一个共价闭合环状DNA,并与结合在ZMW底部的聚合酶结合进行新链合成。这种环状结构的设计能使聚合酶在不间断的情况下持续进行新链合成,其产物也与天然产物一致,因此将产生的这种长读长的序列称为CLR(a continuous long read),这种连续测序模式称为CCS(circular consensus sequencing)[3]。在加入4色荧光标记的4种碱基后,根据碱基互补配对的原则,将相应碱基结合至模板链,不同碱基的加入会激发出不同荧光,可根据产生的荧光信息确定碱基类型。
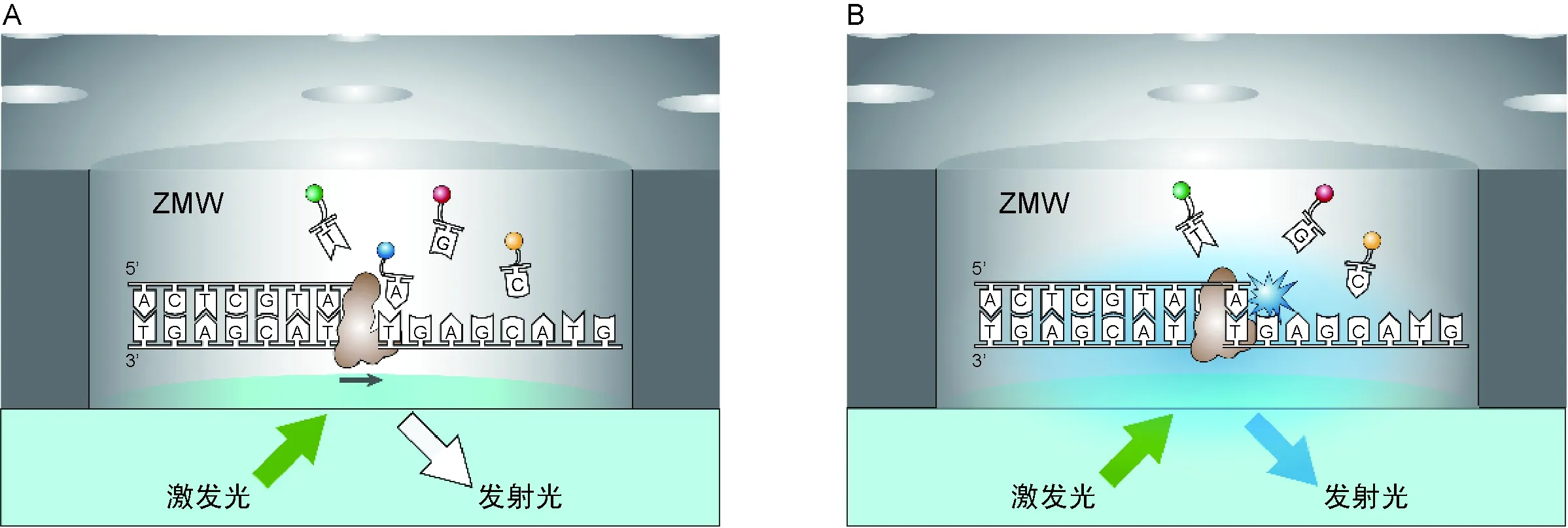
A:4种不同荧光物质标记的核苷酸分别释放入ZMW中(A、T、G和C分别被蓝色、绿色、红色和黄色荧光物质标记),图中显示蓝色荧光物质标记的A被固定于ZMW底部的聚合酶捕获,结合于新链末端;B:在激发光作用下核苷酸上标记的荧光物质发出特异性蓝色荧光,代表所结合的碱基类型。
图1 PacBio测序基本原理图
Fig.1 The schematic diagram of PacBio sequencing
1.2 PacBio测序的特征
PacBio测序采用SMRT原理,最大特点是读长较长,对基因组中一些串联重复区域的检测验证和定位具有独特优势,但出错率较高,且具有随机性。PacBio测序的通量不高,低于Roche454和Illumina平台。此外,PacBio测序平台可通过复制过程中聚合酶的动力学变化特征直接检测碱基的修饰类型和位点信息,为表观遗传学研究提供了新的便捷途径。以下介绍PacBio测序的主要优缺点。
1.2.1 读长较长 最新的PacBio RS Ⅱ测序平台的平均读长已提高至10~15 kb,最大读长可达64.5 kb[4]。Rhoads等利用PacBio RS Ⅱ平台对大小为20 kb的大肠埃希菌DNA进行测序,获得大量不同长度的reads,有报道称PacBio测序平台的平均读长可达10~15 kb[3]。虽然目前尚未有PacBio应用于乙型肝炎病毒(hepatitis B virus,HBV)基因组研究的报道,但该研究表明PacBio的读长已超过HBV基因组全长(3.2 kb),可用于HBV全基因组研究。
1.2.2 可检测碱基修饰 PacBio测序过程中,不同碱基的加入会产生各自特征性的矩形脉冲波,为方便研究,将脉冲时间宽度称为PW(pulse width),相邻两个脉冲波之间的时间宽度称为IPD(interpulse duration)[5]。其中IPD的宽度与碱基修饰关系密切,研究表明存在修饰的碱基通过聚合酶的时间相应延长,在脉冲谱上表现为IPD延长,借以判断序列中碱基修饰数量和位置。此外,不同碱基的修饰类型和相同碱基的不同修饰类型如甲基化等[5]在脉冲波谱上的表示形式不同。亚硫酸氢盐(bisulfite)法是目前最常用的检测DNA甲基化的方法,与之相比,PacBio测序不仅可明显提高检测速度,还可提供不同碱基的修饰信息。
1.2.3 准确率有待提高 PacBio SMRT最明显的缺点是单核苷酸检测的准确率不高,为85%~89%[6]。原因可能是在测序过程中某些特异性荧光信号较弱,易被背景荧光干扰。但这种错误是随机的,可通过提高检测覆盖度来提高测序准确率。当CCS的覆盖度(coverage)达15×时,PacBio测序的准确率高达99.99%[4]。
由于NGS技术具有高通量、高准确率的特点,与PacBio SMRT的读长优势互补,联合使用可大大提高PacBio SMRT的准确率。Tombácz等研究假狂犬病病毒(pseudorabies virus,PRV)的DNA转录情况,采用Illumina和PacBio RS Ⅱ平台分别对PRV的CTO-S(highly abundant short transcript)和CTO-L(less abundant long RNA molecule)进行测序,发现PacBio检测片段长度<700 bp的CTO-S灵敏度不够,只能对片段较长的CTO-L(>2 kb)进行检测;而Illumina平台对较短的CTO-S进行测序更具优势。因此,PacBio的长读长可与Illumina的高通量、高准确率明显优势互补[7]。
1.2.4 通量不高 与NGS技术相比,PacBio测序在通量方面明显不足。如Roche454 GS FLX+的通量为1×106reads/run,Illumina HiSeq 2500(quick run)的通量为1.2×109reads/run(paired),而PacBio RS Ⅱ P6-C4的通量只有(3.5~7.5)×104reads/run[4]。理论上,PacBio的通量应是目前实际通量的2~5倍,因为每个SMRT cell中含有15万个ZMW,在所有导管均可正常测序的情况下,其通量相当可观。但在实际运行过程中,只有3.5万~7万个ZMW可成功完成测序。2015年7月,PacBio公司宣布新型Sequel测序系统将SMRT cell中ZMW从15万个增至100万个,每个SMRT cell的通量提高7倍,同时每次运行的SMRT cell数量提高至16个,极大提高了PacBio测序平台的通量。
2 TGS技术在病毒基因组测序中的应用
目前对病毒基因组的研究主要采用NGS技术,但该技术读长短,无法实现大片段测序,同时存在错误率较高、数据量大、分析困难等问题,限制了其在病毒基因测序方面的应用。TGS技术则与NGS技术形成互补,其读长较长的特点使其在病毒全基因组测序和分析方面具有明显优势。但由于TGS技术市场化较晚,目前应用其进行病毒基因组测序的报道较少,尚有很大应用空间。以下就TGS技术在病毒研究中的应用及其潜在用途进行介绍。
2.1 病毒耐药位点检测
临床上,HBV、丙型肝炎病毒(hepatitis C virus,HCV)和人类免疫缺陷病毒(human immunodeficiency virus,HIV)与某些慢性疾病的发生密切相关,在抗病毒治疗过程中易产生耐药相关突变,影响治疗效果,从而使病毒在宿主体内持续存在,造成疾病慢性化。因此,对病毒耐药突变的检测至关重要。目前最常用的检测方法是直接测序法,灵敏度较低,只能检测出丰度>20%~25%的耐药突变。新兴高通量NGS技术逐渐应用于耐药突变检测,对丰度<5%的突变株具有较高的灵敏度,极大提高了病毒单个位点突变和多重耐药突变的检出率,对研究病毒在机体内的进化特征和指导临床用药具有重要作用[8]。Bergfors等利用PacBio RS Ⅱ平台对HCV相关耐药基因NS5A进行测序,同时对PacBio SMRT技术的性能进行验证[9]。该研究中,Bergfors等用3份GT1a Q3OH、Y93N和GT3a Y93H阳性标本,5份GT1a阳性标本,2份GT3a阴性标本及4种不同稀释倍数的HCV GT1a H17质粒对PacBio SMRT进行验证,并以测序过程的出错率和检测限(limit of detection,LOD)作为性能指标。结果显示,在4种不同稀释倍数的HCV GT1a H17质粒中,平均错误率仅为0.05%~0.25%,可检测频率低至0.24%的NS5A耐药相关变异(resistance-associated variant,RAV)。PacBio SMRT检测NS5A RAV的高灵敏度,可为临床上选择最佳的直接抗病毒药物(direct-acting antiviral,DAA)提供可靠依据,减少因治疗不当而产生的临床耐药。
2.2 病毒基因整合检测
病毒可将自身基因组整合至宿主染色体中而逃避宿主的免疫攻击,实现长期感染,给临床上病毒感染性疾病的诊断、治疗和预后判断带来极大困难。因此,准确检测病毒基因整合位点及其分布特点,可为病毒感染性疾病的治疗提供一定参考。腺相关病毒(adeno-associated virus,AAV)可将自身基因组整合至宿主细胞内,不仅可逃避宿主的免疫攻击,还可导致宿主细胞的癌变。为研究AAV基因组整合的机制,Hüser等利用AAV2作为研究对象。该病毒主要通过调节蛋白Rep78/68结合AAV-ITR(AAV-inverted terminal repeat)内部的RBS(Rep-binding site),将自身基因组整合至宿主AAV S1基因中,同时AAV-ITR也是病毒基因组复制和转录表达的起始位点,在病毒的生命周期中具有重要作用。Hüser等[10]利用PacBio测序平台研究AAV基因组整合至HeLa细胞基因组中的确切位置,发现PacBio SMRT的CCS连续测序模式可实现对单个核苷酸分子多次重复检测,且可对AAV基因组整合位点进行双向检测,极大提高了检测准确率。此外,PacBio测序的长读长不仅实现了AAV全基因组的覆盖,还可延伸至目的序列以外区域,对AAV基因组整合位点的确定至关重要。
2.3 宏病毒组测序
病毒宏基因组学(metagenomics)的研究对象是特定环境中的整个病毒群体,利用基因测序技术和泛病毒芯片技术获取并集合环境中全部病毒的基因信息,对新病毒的发现、病原体溯源和病毒感染预警具有重要意义[11]。病毒基因组学突破传统的病毒学检测方法〔如血清免疫学、电镜、聚合酶链反应(polymerase chain reaction,PCR)和细胞培养等〕,利用泛病毒芯片和基因测序技术检测病毒。但采用泛病毒芯片技术对序列未知的、环境中丰度<5%和变异程度较大的病毒较难检测。最近几年NGS技术在病毒宏基因组学研究中的应用越来越广泛,对新病毒的发现和病毒变异检测具有重要作用。2010年,Donaldson等分析蝙蝠肠道内的病毒构成,共获得近60万个片段的核酸序列,经组装发现这些片段来自3组以上新的1型冠状病毒、不同昆虫及2种新型细菌的质粒基因组序列。该研究表明,蝙蝠体内携带多种不同病毒,并可传播给自然界中其他宿主[12]。Greninger等利用超深度测序和病毒基因芯片技术研究甲型流感病毒H1N1时,获取了大量新变异株,同时发现了许多未知新病毒,表明超深度测序可能取代传统的病毒鉴定方法,更好地用于临床上未知疾病的诊断和新病毒的检测[13]。Willner等采用病毒宏基因组学对5例肺囊性纤维化患者和5例正常健康者痰液中的病毒进行差异性研究,发现肺囊性纤维化患者痰液中病毒群体相似度较高,而与对照组的差异较明显;同时还发现在肺囊性纤维化的不同病程阶段,患者体内的芳香族氨基酸代谢方式有所不同。该研究提示,可通过改变患者机体内的微生物群落达到治疗疾病的目的[14]。这些数据均是通过NGS技术获得。目前,新兴的TGS技术尚未用于相关研究,但凭借其优势,相信以后也可用于病毒宏基因组学的研究并带来新的研究策略。
2.4 病毒准种研究
病毒准种(quasispecies)的概念由Eigen和Schuster于1977年提出,是指受到遗传变异、竞争及环境选择作用等影响而形成的高度相关但又不完全相同的变异株和重组基因组组成的动态种群[15]。HIV、HBV和HCV等病毒的聚合酶缺乏对新合成核苷酸链的修饰作用,使基因在复制过程中产生较高的错配率。例如,HBV的错配率为10-4/核苷酸,每天产生的错配核苷酸有1010个[16]。目前准种研究主要采用克隆-测序技术,该方法费时、费力,在克隆过程中会引入偏倚,限制了病毒异质性的分析。此外,NGS技术在文库构建过程中也用到PCR扩增,导致测序结果出现插入和缺失的假阴性结果。TGS技术可省去扩增过程,避免引入人为错误,从而提高对病毒微变异株的检出率。Bull等[17]等分别利用Illumina NGS平台和PacBio TGS平台对122例HCV感染患者进行HCV全长准种测序,结果发现虽然Illumina平台测序深度高,易检测多种基因型的共感染,但PacBio平台平均扩增长度达9 kb,使检测同一病毒株中的联合变异成为可能。Huang等[18]利用Illumina Miseq平台和PacBio RS Ⅱ平台对人工构建的HIV准种进行测序比较,结果发现PacBio可检测频率低至1%的变异,特别是联合变异,能为临床检测耐药突变提供更多信息。相信TGS技术凭借其明显的优势,在病毒准种研究方面可弥补NGS技术的不足。
3 结语
随着人类对基因的研究越来越深入,对基因测序技术也提出了更高的要求。以PacBio SMRT为代表的TGS技术与NGS技术相比,最明显的优势是弥补了后者读长短的缺陷,但随之带来的是测序错误率较高和成本提升。若将TGS技术广泛用于病毒变异研究,首先要提高该技术的准确率,并降低检测成本,这样才能更好地用于临床上病毒感染性疾病的监测,为临床抗病毒治疗提供相应参考。
除目前已商品化的NGS和TGS技术,基于电子传导技术进行碱基检测的第四代测序技术已日渐成熟。第四代测序技术与TGS技术相比,可避免洗脱和扩增过程中造成的误差,且具有超长读长、高通量和更短测序时间等特点[19]。总之,不同的测序平台有各自优缺点,突飞猛进的测序技术可为临床检测提供更多的选择,也极大促进了临床医学的进步。
[1] 张拥军.高通量测序技术在临床病毒学领域的应用 [J].中国人兽共患病学报,2015,31(9):864-869, 875.
[2] Miyamoto M, Motooka D, Gotoh K, Imai T, Yoshitake K, Goto N, Iida T, Yasunaga T, Horii T, Arakawa K, Kasahara M, Nakamura S. Performance comparison of second- and third-generation sequencers using a bacterial genome with two chromosomes [J]. BMC Genomics, 2014, 15: 699. doi: 10.1186/1471-2164-15-699.
[3] Rhoads A, Au KF. PacBio sequencing and its applications [J]. Genomics Proteomics Bioinformatics, 2015, 13(5): 278-289.
[4] Eid J, Fehr A, Gray J, Luong K, Lyle J, Otto G, Peluso P, Rank D, Baybayan P, Bettman B, Bibillo A, Bjornson K, Chaudhuri B, Christians F, Cicero R, Clark S, Dalal R, Dewinter A, Dixon J, Foquet M, Gaertner A, Hardenbol P, Heiner C, Hester K, Holden D, Kearns G, Kong X, Kuse R, Lacroix Y, Lin S, Lundquist P, Ma C, Marks P, Maxham M, Murphy D, Park I, Pham T, Phillips M, Roy J, Sebra R, Shen G, Sorenson J, Tomaney A, Travers K, Trulson M, Vieceli J, Wegener J, Wu D, Yang A, Zaccarin D, Zhao P, Zhong F, Korlach J, Turner S. Real-time DNA sequencing from single polymerase molecules [J]. Science, 2009, 323(5910): 133-138.
[5] Schadt EE, Banerjee O, Fang G, Feng Z, Wong WH, Zhang X, Kislyuk A, Clark TA, Luong K, Keren-Paz A, Chess A, Kumar V, Chen-Plotkin A, Sondheimer N, Korlach J, Kasarskis A. Modeling kinetic rate variation in third generation DNA sequencing data to detect putative modifications to DNA bases [J]. Genome Res, 2013, 23(1): 129-141.
[6] 张欣欣, 王铭杰. 新一代测序技术在HBV变异研究中的应用 [J]. 临床肝胆病杂志, 2015, 31(4): 514-519.
[7] Tombácz D, Csabai Z, Oláh P, Havelda Z, Sharon D, Snyder M, Boldogköi Z. Characterization of novel transcripts in pseudorabies virus [J]. Viruses, 2015, 7(5): 2727-2744.
[8] 龚玲, 张欣欣. 高通量测序技术在病毒感染性疾病研究中的应用 [J]. 中华传染病杂志, 2013, 31(3): 187-189.
[9] Bergfors A, Leenheer D, Bergqvist A, Ameur A, Lennerstrand J. Analysis of hepatitis C NS5A resistance associated polymorphisms using ultra deep single molecule real time (SMRT) sequencing [J]. Antiviral Res, 2016, 126: 81-89.
[10] Hüser D, Gogol-Döring A, Chen W, Heilbronn R. Adeno-associated virus type 2 wild-type and vector-mediated genomic integration profiles of human diploid fibroblasts analyzed by third-generation PacBio DNA sequencing [J]. J Virol, 2014, 88(19): 11253-11263.
[11] 何彪, 涂长春. 病毒宏基因组学的研究现状及应用 [J]. 畜牧兽医学报, 2012, 43(12): 1865-1870.
[12] Donaldson EF, Haskew AN, Gates JE, Huynh J, Moore CJ, Frieman MB. Metagenomic analysis of the viromes of three North American bat species: viral diversity among different bat species that share a common habitat [J]. J Virol, 2010, 84(24): 13004-13018.
[13] Greninger AL, Chen EC, Sittler T, Scheinerman A, Roubinian N, Yu G, Kim E, Pillai DR, Guyard C, Mazzulli T, Isa P, Arias CF, Hackett J, Schochetman G, Miller S, Tang P, Chiu CY. A metagenomic analysis of pandemic influenza A (2009 H1N1) infection in patients from North America [J]. PLoS One, 2010, 5(10): e13381.
[14] Willner D, Furlan M, Haynes M, Schmieder R, Angly FE, Silva J, Tammadoni S, Nosrat B, Conrad D, Rohwer F. Metagenomic analysis of respiratory tract DNA viral communities in cystic fibrosis and non-cystic fibrosis individuals [J]. PLoS One, 2009, 4(10): e7370.
[15] Domingo E, Gomez J. Quasispecies and its impact on viral hepatitis [J]. Virus Res, 2007, 127(2): 131-150.
[16] Dahari H, Shudo E, Ribeiro RM, Perelson AS. Modeling complex decay profiles of hepatitis B virus during antiviral therapy [J]. Hepatology, 2009, 49(1): 32-38.
[17] Bull RA, Eltahla AA, Rodrigo C, Koekkoek SM, Walker M, Pirozyan MR, Betz-Stablein B, Toepfer A, Laird M, Oh S, Heiner C, Maher L, Schinkel J, Lloyd AR, Luciani F. A method for near full-length amplification and sequencing for six hepatitis C virus genotypes [J]. BMC Genomics, 2016, 17: 247.
[18] Huang DW, Raley C, Jiang MK, Zheng X, Liang D, Rehman MT, Highbarger HC, Jiao X, Sherman B, Ma L, Chen X, Skelly T, Troyer J, Stephens R, Imamichi T, Pau A, Lempicki RA, Tran B, Nissley D, Lane HC, Dewar RL. Towards better precision medicine: PacBio single-molecule long reads resolve the interpretation of HIV drug resistant mutation profiles at explicit quasispecies (haplotype) level [J]. J Data Mining Genomics Proteomics, 2016, 7(1):182.doi: 10.4172/2153-0602.1000182.
[19] Ke R, Mignardi M, Hauling T, Nilsson M. Fourth generation of next-generation sequencing technologies: promise and consequences [J/OL]. Hum Mutat, 2016 [Epub ahead of print]. http://onlinelibrary.wiley.com/doi/10.1002/humu.23051/epdf.
. ZHANG Xinxin,E-mail: zhangxinxinrj@163.com
Characteristics of the third generation sequencing technology and its application in researching of viral genomes
YUAN Yi, WANG Mingjie, ZHANG Xinxin
ResearchUnitofClinicalVirology,RuijinHospital,ShanghaiJiaoTongUniversitySchoolofMedicine,Shanghai200025,China
With the consecutive innovation and application of gene sequencing technology, the third generation sequencing (TGS) based on single molecule real time (SMRT) sequencing technology has demonstrated a potential in a variety of applications in basic and clinical studies, including whole genome assembly, detection of gene structure variation, epigenetic research, etc. This review mainly introduces the principle, characteristics and applications of the TGS technology, especially its applications in virus researches. This review also compares the differences between the next generation sequencing (NGS) and TGS, so as to provide a reference for application based selection for sequencing technology.
Third generation sequencing; Pacific Biosciences; Single molecule real time sequencing; Viral genome
国家“十二五”科技重大专项(2012ZX10002007)
张欣欣
2016-06-13)
猜你喜欢
农业灾害研究(2022年1期)2022-05-07 01:31:04
今日农业(2021年11期)2021-08-13 08:53:24
教学考试(高考生物)(2020年6期)2020-11-23 05:25:56
食品与生物技术学报(2020年8期)2020-01-06 08:00:56
科学24小时(2019年5期)2019-06-11 08:39:38
发明与创新(2019年9期)2019-03-26 02:22:48
化工进展(2015年6期)2015-11-13 00:26:29
遗传(2014年3期)2014-02-28 20:58:49
世界科学(2014年8期)2014-02-28 14:58:31
中国海洋大学学报(自然科学版)(2014年12期)2014-02-28 12:22:06
