
水痘-带状疱疹病毒潜伏感染的研究现状
2017-01-05 11:00:26汤华民贾俊丽姚堃
微生物与感染 2016年6期
汤华民,贾俊丽,姚堃
南京医科大学免疫学系,南京 211166
·综述·
水痘-带状疱疹病毒潜伏感染的研究现状
汤华民,贾俊丽,姚堃
南京医科大学免疫学系,南京 211166
水痘-带状疱疹病毒潜伏感染后的再活化可引起带状疱疹,其潜伏感染与再活化的机制还不完全清楚。本文综述了该病毒潜伏感染和再活化的实验模型新进展,并概述了从这些实验模型中所获得的最新知识。
水痘-带状疱疹病毒;潜伏感染;动物模型
水痘-带状疱疹病毒(varicella-zoster virus,VZV)和单纯疱疹病毒(herpes simplex virus,HSV)同属疱疹病毒α亚科,具有嗜神经性。VZV的初次感染主要发生在儿童,可引起水痘,水痘愈后VZV长期潜伏于宿主神经节中。随着宿主年龄增加或免疫力低下时,VZV会再次活化,引起带状疱疹。水痘疫苗对水痘发病有很好的预防作用;对于带状疱疹的预防,目前只有利用高效价水痘疫苗开发而成的带状疱疹疫苗,但带状疱疹疫苗仅在美国使用,尚未在世界范围内推广。在高龄化进程不断加速的今天,高效价带状疱疹疫苗的开发与推广有重要的社会意义。本文结合当前VZV研究的热点,重点综述VZV潜伏感染及再活化的最新进展。
1 VZV的感染周期
1.1 VZV的入侵与扩散
VZV主要通过飞沫经呼吸道或直接接触局部黏膜进入机体后增殖而传播。有关VZV如何进入细胞,到目前为止了解并不多;而HSV入侵细胞的过程已解析得较清楚。HSV入侵细胞时,病毒表面的糖蛋白D(glycoprotein D,gD)与细胞表面的特异性受体相互作用而进入细胞。可通过病毒包膜与细胞膜直接融合进入细胞(非pH依赖细胞入侵过程);或通过细胞内吞作用进入细胞,然后病毒包膜与细胞内膜融合从而释放病毒核衣壳(pH依赖细胞入侵过程)。VZV颗粒中不表达HSV gD的同源糖蛋白,但其包膜中表达大量gE。曾有报道[1]称细胞内的胰岛素降解酶(insulin-degrading enzyme,IDE)能与gE结合,由此认为IDE可能是VZV入侵细胞的受体;然而,进一步实验证明IDE主要与未成熟gE相结合。因此,IDE是否为VZV入侵细胞的受体仍需验证[2]。此外,因为VZV的糖蛋白富含甘露糖6磷酸(mannose 6-phosphate,M6P),所以VZV可能是通过M6P与M6P受体(mannose 6-phosphate receptor,MPR)结合而进入细胞,此假说还需进一步证实[3]。VZV入侵细胞后,其核衣壳释放至细胞质,然后转运至细胞核膜,其核酸释放至细胞核内,随后病毒开始复制。新合成的病毒核衣壳从细胞核内膜获得初级包膜(primary envelope),进而进入细胞核的内膜与外膜之间。接着,病毒的初级包膜与细胞核的外膜融合,病毒的核衣壳释放至细胞质,细胞质中的核衣壳从反面高尔基体管网状结构(trans-Golgi network,TGN)或TGN来源的细胞内膜获得次级包膜(secondary envelope)而包装成熟,然后成熟的病毒颗粒释放至细胞外,感染新的细胞。入侵机体的VZV先在局部黏膜组织中增殖,然后向周围淋巴组织传播、扩散。淋巴组织中被感染的T细胞进入血液,进而向皮肤组织传播、扩散,临床上呈现水痘。一般从VZV感染黏膜组织至水痘出现需10~21 d,这段时间可被认为是病毒感染T细胞,T细胞再感染皮肤组织所需的时间。VZV感染的T细胞主要为记忆性T细胞,表达T细胞活化抗原和归巢抗原,由此推断感染皮肤组织的VZV来源于感染的T细胞[4]。此外,被VZV感染的T细胞表达信号转导与转录激活因子3(signal transducer and activator of transcription 3,STAT3)和生存素(survivin)等蛋白,具有抗细胞凋亡的功能,从而保证病毒从T细胞向皮肤组织传播的所需时间[5]。
1.2 VZV的神经系统感染
皮肤组织中感染、增殖的VZV会沿神经系统的轴突逆行输送至神经元的细胞体部,进而在此形成潜伏感染(图1A)。潜伏感染的病毒在宿主高龄和免疫抑制时会再次活化,通过顺向运输到达并感染皮肤组织,引起带状疱疹(图1B)。
1.3 VZV感染相关蛋白
从VZV体内感染过程来看,其具有嗜上皮、嗜T细胞和嗜神经性的特性,这些特性也被实验证实。VZV感染T细胞时,很难观察到病毒引起的细胞融合;而当VZV感染上皮细胞或神经细胞时,细胞融合非常明显。神经系统细胞表面表达髓磷脂相关糖蛋白(myelin-associated glycoprotein,MAG),VZV包膜表面表达的gB糖蛋白能与MAG结合,对VZV入侵神经细胞及病毒引起神经细胞融合起关键作用。但MAG在VZV感染其他细胞(如上皮细胞)中的作用还有待研究。VZV感染上述3种细胞时,均需gE参与。此外,在VZV感染上皮细胞和神经细胞时还需病毒gI参与。gE与gI能相互作用,并以糖蛋白复合物的形式参与病毒入侵细胞的过程。
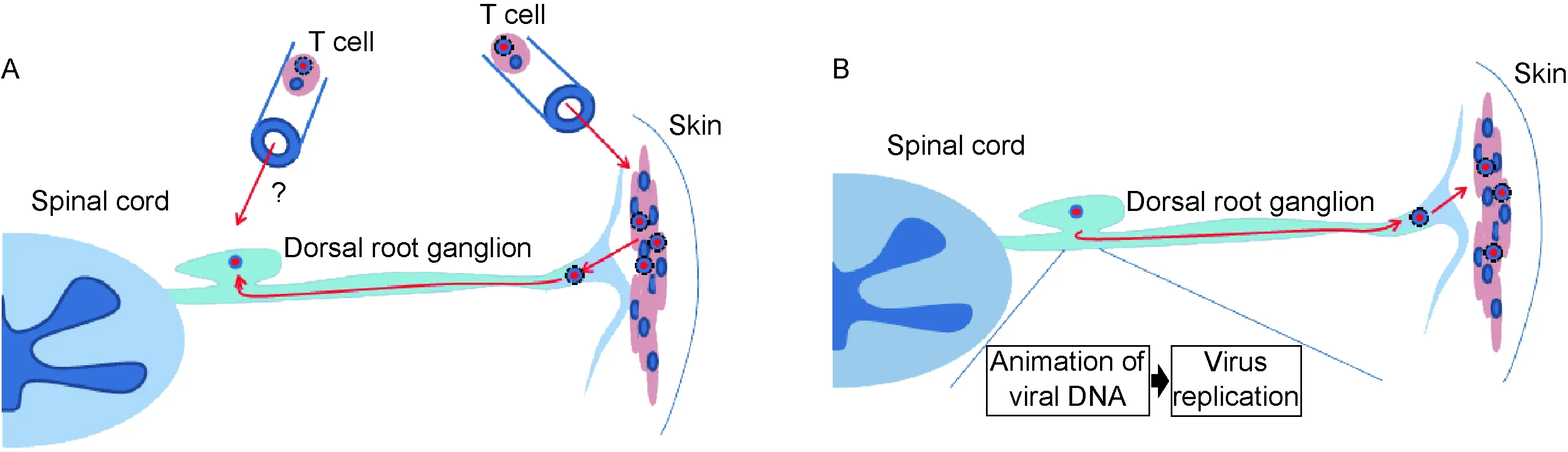
A: VZV-infected T cells transmit the virus to skin tissue where the virus productively replicates and gains the access to the neuron through retrograde transport. Recently, it has also been reported that the virus might get to the neuron by the transmission of T cells. B: After reactivation, the VZV particles return back to the skin tissue through anterograde transport.
图1 VZV潜伏感染的建立与再活化
Fig.1 Establishment and reactivation of VZV latent infection
2 VZV潜伏感染的实验模型
在HSV研究中,已建立潜伏感染小鼠、家兔及培养细胞模型。由于VZV感染局限于人,故利用动物模型来研究其潜伏感染非常困难。到目前为止,关于VZV潜伏感染的结论主要来自尸体解剖样品的研究。虽然有文献报道利用小鼠、家兔等小型动物来研究VZV感染现象,但尚未能建立人体中VZV潜伏感染再活化的动物模型[6-7]。
2.1 VZV潜伏感染豚鼠模型
20多年前,有团队利用豚鼠培养细胞驯化VZV,并利用驯化的病毒感染豚鼠,从而研究VZV感染引起的病理损伤等现象,但没有研究被驯化病毒感染的豚鼠感觉性神经节中是否存在病毒核酸,或是否有病毒基因产物表达,因此该病毒是否能在豚鼠中建立潜伏感染还不清楚[8-9]。近年来,Gan等发现VZV感染能引起人肠道病变,并从肠道组织中检出VZV DNA。基于此,该研究团队利用VZV感染的人外周血单个核细胞(peripheral blood mononuclear cell,PBMC)来感染豚鼠,从而建立VZV豚鼠肠管神经节潜伏感染模型,但该模型中潜伏感染的病毒是否可再次活化还不清楚[10]。有趣的是,体外实验利用豚鼠的肠道神经节可建立VZV潜伏感染和再活化模型,解析了潜伏感染病毒基因的表达[11],但该模型是否能再现人肠道神经节感染还有待进一步研究。
2.2 人源化严重联合免疫缺陷(severe combined immunodeficiency-humanized,SCID-hu)小鼠模型
VZV研究中使用的SCID-hu 小鼠模型由Arvin等开发,是在SCID小鼠的基础上移植人的皮肤等组织构建而成。其后,用VZV感染移植的人体组织,可动态研究VZV在人体组织中的感染现象与机制。如果在SCID小鼠中移植了来自终止妊娠胎儿的背根神经节(dorsal root ganglion),再用VZV感染移植组织,就能建立VZV神经感染模型。利用该模型,Arvin等详细研究了VZV在溶解性感染和潜伏感染时的病毒基因产物。然而,该模型中病毒感染神经组织不是血源或由上皮顺向运输而来,而是通过细胞-细胞间感染或直接利用病毒颗粒(cell-free virus)感染,因此其能否再现人体内的潜伏感染还有待研究。
2.3 VZV感染灵长类动物模型
VZV感染侏狨等灵长类动物时,只能看到临床症状不明显的非显性感染,且尚未能确认这些感染动物中的VZV潜伏感染。因此,利用灵长类动物研究VZV潜伏感染时,可能需使用与人类更接近的、更高等的灵长类动物。
2.4 基于猴带状疱疹病毒(simian varicella virus,SVV)的动物模型
SVV核酸序列与VZV约有70%的同源性,在很大程度上SVV感染与VZV感染非常相像。SVV感染猴后,引起病毒血症,呈现水痘症状,动物体内能检测到T、B细胞免疫反应,且能形成潜伏感染,潜伏感染的病毒亦能再次活化(因使用的猴种类及病毒感染方式不一样,有时只能看到其中一部分现象)。需强调的是,如果对SVV潜伏感染的猴给予免疫抑制剂,时常能检测到病毒再活化,且出现带状疱疹症状。由此可见,SVV动物模型是研究VZV类病毒较为有用的动物模型。然而,对SVV感染的猴神经组织进行详细分析后发现,与HSV潜伏感染一样,在SVV感染的猴神经组织中检测到潜伏感染相关转录体(latency-associated transcript,LAT)的产物。虽然对LAT在病毒潜伏感染中的作用还不完全清楚,但到目前为止还没有在VZV潜伏感染的人神经组织中检测到LAT表达。因此,SVV与VZV维持潜伏感染的机制有可能不同。
2.5 分化神经细胞模型
近年,利用iPS细胞或ES细胞分化来的神经细胞进行VZV神经细胞感染机制研究成为可能。如果用高感染复数(multiplicity of infection,MOI)病毒来感染这些细胞,细胞出现病变而死亡;如果用低MOI病毒来感染这些细胞,细胞能存活2周以上,且不出现明显的细胞病变[12]。进一步研究表明,VZV对这些细胞会呈现不明显感染[13]。高通量测序研究表明,在VZV感染的神经细胞中病毒基因的表达量与该病毒在成纤维细胞感染中的基因量相比,只有12个病毒基因表达有显著差异。低MOI VZV感染的神经细胞不出现病变很有可能是VZV DNA数量少[14]。利用该感染系统研究VZV在神经细胞中的运输也将成为可能。此外,如果将分化的神经细胞进行3D培养,这些细胞能存活6个月以上,从而使研究VZV的持续感染成为可能。
3 VZV的潜伏感染与活化
3.1 VZV潜伏感染的确立
一般认为,初次感染时VZV在皮肤组织中增殖后向神经细胞传播,该过程中VZV通过逆行运输到达神经细胞的细胞体。非常有趣的是,在SVV感染研究中,皮肤出现疱疹前已能在神经细胞的细胞体部检测到该病毒[15]。VZV能感染记忆性T细胞[16]。在利用SCID-hu小鼠的VZV感染实验中发现,VZV感染的记忆性T细胞能向神经节传播病毒[17]。由此可见,VZV除从皮肤组织向神经组织传播外,也可能通过感染的T细胞向神经组织传播(图1A)。
从病毒到达神经细胞的细胞体至病毒潜伏感染建立是否需病毒增殖,还不是很清楚。在HSV研究中,一般被接受的是“多次感染”(round trip infection)的神经系统感染模型。该模型中,病毒到达神经细胞的细胞体后先增殖,再通过顺行运输感染皮肤组织,病毒在皮肤组织再次增殖后通过逆行运输向神经系统传播,这样病毒就能感染更多的神经细胞,但病毒增殖对其潜伏感染的建立不是必需的[18]。关于VZV是否通过同样的感染机制来建立潜伏感染,是今后的研究方向。
VZV核酸进入细胞核后,主要被早幼粒细胞性白血病(promyelocytic leukemia,PML)蛋白和核结构域10(nuclear domain 10,ND10)蛋白组成的PML/ND10核小体所识别。PML/ND10核小体的抗病毒作用至少有两种机制:其一,PML/ND10核小体作用于病毒核酸启动子区域,使之异染色质(heterochromatin)化,导致病毒基因沉默[19-20]。其二,PML/ND10核小体将病毒核酸局限于核内一定区域,也能抑制病毒增殖[21]。此外,游离的PML/ND10复合体会形成PML的笼状结构(cage),将新合成的病毒核衣壳局限于核内一定区域,也有助于病毒感染的沉默[21]。在VZV感染细胞中,VZV能通过即刻早期蛋白61(immediate-early protein 61,IE61)阻碍PML/ND10核小体功能,此过程中PML/ND10核小体结构遭到破坏,但其蛋白一般不被降解,且VZV感染后IE61的抗PML/ND10核小体功能是一过性的。PML/ND10核小体及其复合物是否在VZV神经细胞潜伏感染的确立中起关键作用,也是今后的研究方向。
3.2 VZV潜伏感染时病毒基因的表达及病毒潜伏感染的维持
研究尸体解剖获得的人神经细胞结果显示,在人三叉神经中能检测到的VZV DNA数量为101~104拷贝/100 ng总DNA。此外,只能检测到为数不多的病毒基因产物。到目前为止,能检出的病毒基因产物有:ORF4、ORF11、ORF18、ORF29、ORF40、ORF41、ORF43、ORF57、ORF62、ORF63、ORF66、ORF68[22-24]。有趣的是,从死亡到尸体解剖的时间影响病毒基因产物的检测,尸体解剖越早,检出的病毒基因产物越少。因此,目前检出的病毒基因产物是否是真正的病毒潜伏感染时的基因产物,或只是死亡导致的神经病变引起的病毒基因表达产物,还需进一步研究[25]。
死后24 h内的尸体解剖神经节结果表明,病毒核酸的ORF62与ORF63基因启动子区域被常染色质(euchromatin)化,这些基因的表达及其功能可能有助于病毒潜伏感染的维持[26]。病毒ORF63基因产物在死后9 h内的尸体解剖样品中亦能检出,且是检出最多的病毒基因产物。VZV溶解性感染时,ORF63的产物(IE63)以磷酸化形式存在于细胞核内[27-28],并与人抗沉默功能蛋白1(antisilencing function 1,ASF1)结合,该复合物能调节病毒的溶解性感染与潜伏感染[29]。此外,IE63也能抑制病毒感染引起的机体干扰素分泌[30-31]。IE63可能对病毒潜伏感染的建立与维持起重要作用。然而到目前为止,对IE63功能的解析主要是利用上皮细胞而不是神经细胞,因此其在病毒感染神经细胞中的作用需进一步研究。目前,尚无研究报道VZV ORF62基因在病毒潜伏感染中的功能。
3.3 VZV的活化
潜伏感染的VZV在高龄者机体处于免疫抑制时会再次活化,其分子水平的活化机制尚不完全清楚。VZV再活化机制的研究是当今病毒研究的热点之一。
HSV和VZV可感染同一神经节、同一神经节中相邻的神经细胞或同一神经细胞而建立潜伏感染,但两种病毒的再活化情况不一样。HSV再活化引起的疾病主要在年轻人中多见,且在同一患者中可反复再活化、致病。VZV再活化引起的带状疱疹多见于老年人群,一般只发病一次[32]。由此可见,两种病毒的再活化机制很有可能是不同的。因为HSV潜伏动物模型已建立,其再活化研究的进展非常迅速;而对于VZV再活化,因为没有合适的动物模型,研究进展比较缓慢。
尸体解剖中的VZV再活化,很可能是组织处于低氧状况造成的,低氧对神经细胞造成压力(stress),进而导致VZV再活化。低氧情况下,细胞内下调蛋白表达的哺乳动物雷帕霉素靶蛋白(mammalian target of rapamycin,mTOR)功能被抑制,使病毒蛋白合成抑制被解除,从而诱导病毒再活化[33]。在其他α疱疹病毒研究中,除低氧环境外,还可利用药物等方法来诱导病毒再活化。在这些实验中,通过某些处理诱导细胞凋亡信号和某些细胞因子表达可促进病毒再活化[34-35]。
在VZV再活化模型中,因为神经细胞压力等因素而造成病毒核酸活化是病毒活化的第1步。潜伏感染的病毒核酸受异染色质与常染色质修饰而控制病毒基因的表达。两种核酸修饰部位因为有CTCF(CCCTC-binding factor)的存在而被隔开。病毒核酸再活化时,CTCF会从病毒核酸上脱落,造成核酸修饰改变,进而导致病毒再活化。在HSV研究中,活化的病毒核酸基因表达不是遵循疱疹病毒初次感染时的病毒基因表达顺行〔经典级联(classical cascade):早早期基因——早期基因——晚期基因〕,而是各期基因一起表达。此外,这些基因的表达也不依赖于某个病毒蛋白的表达或病毒核酸的复制[36]。尸体解剖神经节的VZV研究结果表明,随着死亡后时间的推移,病毒基因表达数目增加,且与HSV一样,VZV基因表达不遵循经典级联。如果在病毒活化阶段消除诱导神经细胞压力的因素,再活化的病毒核酸就能再次沉默。如果细胞压力持续存在,病毒基因就能大量表达,最终形成经典级联的病毒基因表达。其后,新的病毒颗粒被组装,病毒从神经组织向皮肤组织传播并感染,导致带状疱疹发生。
利用SCID-hu小鼠来研究HSV和VZV的神经组织感染发现,HSV感染神经组织中观察不到细胞融合;而在VZV感染神经组织中,VZV不仅感染神经元,还能感染其周围的卫星细胞,可见到细胞融合形成的多核巨细胞,且观察到的VZV感染现象在尸体解剖组织中也能观察到[37-38]。这些感染现象很可能与带状疱疹引起的剧烈神经疼痛相一致[39]。
4 结语
与HSV相比,VZV潜伏感染研究进展非常缓慢,原因之一是还没有建立VZV潜伏感染与再活化动物模型。VZV潜伏感染动物模型的构建将是VZV研究的一个重点课题,不仅有助于VZV潜伏感染与再活化机制的研究,还有助于控制病毒再活化方法的开发。最近,利用iPS或ES细胞分化神经细胞来研究VZV感染机制已成为可能。如果病毒感染方式等能进一步改良,也将有助于VZV潜伏感染机制的研究。
[1] Li QX, Ali MA, Cohen JI. Insulin degrading enzyme is a cellular receptor mediating varicella-zoster virus infection and cell-to-cell spread [J]. Cell, 2006, 127(2): 305-316.
[2] Carpenter JE, Jackson W, de Souza GA, Haarr L. Insulin-degrading enzyme binds to the nonglycosylated precursor of varicella-zoster virus gE protein found in the endoplasmic reticulum [J]. J Virol, 2010, 84(2): 847-855.
[3] Chen JJ, Zhu ZL, Gershon AA, Gershon MD. Mannose 6-phosphate receptor dependence of varicella zoster virus infection in vitro and in the epidermis during varicella and zoster [J]. Cell, 2004, 119(7): 915-926.
[4] Ku CC, Padilla JA, Grose C, Butcher EC, Arvin AM. Tropism of varicella-zoster virus for human tonsillar CD4+T lymphocytes that express activation, memory, and skin homing markers [J]. J Virol, 2002, 76(22): 11425-11433.
[5] Sen N, Che XB, Rajamani J, Zerboni LA, Ptacek J, Arvin AM. Signal transducer and activator of transcription 3 (STAT3) and survivin induction by varicella-zoster virus promote replication and skin pathogenesis [J]. Proc Natl Acad Sci USA, 2012, 109(2): 600-605.
[6] Haberthur K, Messaoudi I. Animal models of varicella zoster virus infection [J]. Pathogens, 2013, 2(2): 364-382.
[7] Kinchington PR, Goins WF. Varicella zoster virus-induced pain and post-herpetic neuralgia in the human host and in rodent animal models [J]. J Neurovirol, 2011, 17(6): 590-599.
[8] Lowry PW, Sabella C, Koropchak CM, Watson BN, Thackray HM, Abbruzzi GM, Arvin AM. Investigation of the pathogenesis of varicella-zoster virus infection in guinea pigs by using polymerase chain reaction [J]. J Infect Dis, 1993, 167(1): 78-83.
[9] Myers MG, Connelly BL, Stanberry LR. Varicella in hairless guinea pigs [J]. J Infect Dis, 1991, 163(4): 746-751.
[10] Gan L, Wang ML, Chen JJ, Gershon MD. Infected peripheral blood mononuclear cells transmit latent varicella zoster virus infection to the guinea pig enteric nervous system [J]. J Neurovirol, 2014, 20(5): 442-456.
[11] Chen JJ, Gershon AA, Li ZS, Lungu O, Gershon MD. Latent and lytic infection of isolated guinea pig enteric ganglia by varicella zoster virus [J]. J Med Virol, 2003, 70(Suppl 1): S71-S78.
[12] Sadaoka T, Depledge DP, Rajbhandari L, Venkatesan A, Breuer J, Cohen JI. In vitro system using human neurons demonstrates that varicella-zoster vaccine virus is impaired for reactivation, but not latency [J]. Proc Natl Acad Sci USA, 2016, 113(17): E2403-E2412.
[13] Grose C, Yu XL, Cohrs RJ, Carpenter JE, Bowlin JL, Gilden D. Aberrant virion assembly and limited glycoprotein C production in varicella-zoster virus-infected neurons [J]. J Virol, 2013, 87(17): 9643-9648.
[14] Baird NL, Bowlin JL, Yu X, JonjiS, Haas J, Cohrs RJ, Gilden D. Varicella zoster virus DNA does not accumulate in infected human neurons [J]. Virology, 2014. doi: 10.1016/j.virol.2014.04.014.
[15] Ouwendijk WJ, Mahalingam R, de Swart RL, Haagmans BL, van Amerongen G, Getu S, Gilden D, Osterhaus AD, Verjans GM. T-cell tropism of simian varicella virus during primary infection [J]. PLoS Pathog, 2013, 9(5): e1003368.
[16] Ku CC, Zerboni L, Ito H, Graham BS, Wallace M, Arvin AM. Varicella-zoster virus transfer to skin by T cells and modulation of viral replication by epidermal cell interferon-alpha [J]. J Exp Med, 2004, 200(7): 917-925.
[17] Zerboni L, Ku CC, Jones CD, Zehnder JL, Arvin AM. Varicella-zoster virus infection of human dorsal root ganglia in vivo [J]. Proc Natl Acad Sci USA, 2005, 102(18): 6490-6495.
[18] Steiner I, Spivack JG, Deshmane SL, Ace CI, Preston CM, Fraser NW. A herpes simplex virus type 1 mutant containing a nontransinducing Vmw65 protein establishes latent infection in vivo in the absence of viral replication and reactivates efficiently from explanted trigeminal ganglia [J]. J Virol, 1990, 64(4): 1630-1638.
[19] Newhart A, Rafalska-Metcalf IU, Yang TA, Janicki SM. Single-cell analysis of Daxx and ATRX-dependent transcriptional repression [J]. J Cell Sci, 2012, 125(22): 5489-5501.
[20] Lukashchuk V, Everett RD. Regulation of ICP0-Null mutant herpes simplex virus type 1 infection by ND10 components ATRX and hDaxx [J]. J Virol, 2010, 84(8): 4026-4040.
[21] Reichelt M, Wang L, Sommer M, Perrino J, Nour AM, Sen N, Baiker A, Zerboni L, Arvin AM. Entrapment of viral capsids in nuclear PML cages is an intrinsic antiviral host defense against varicella-zoster virus [J]. PLoS Pathog, 2011, 7(2): e1001266.
[22] Cohrs RJ, Gilden DH, Kinchington PR, Grinfeld E, Kennedy PG. Varicella-zoster virus gene 66 transcription and translation in latently infected human ganglia [J]. J Virol, 2003, 77(12): 6660-6665.
[23] Kennedy PG, Grinfeld E, Bell JE. Varicella-zoster virus gene expression in latently infected and explanted human ganglia [J]. J Virol, 2000, 74(24): 11893-11898.
[24] Nagel MA, Choe A, Traktinskiy IA, Gilden D, Cohrs RJ. Varicella-zoster virus transcriptome in latently infected human ganglia [J]. J Virol, 2011, 85(5): 2276-2287.
[25] Ouwendijk WJ, Choe A, Nagel MA, Osterhaus AD, Cohrs RJ. Restricted varicella-zoster virus transcription in human trigeminal ganglia obtained soon after death [J]. J Virol, 2012, 86(18): 10203-10206.
[26] Gary L, Gilden DH, Cohrs RJ. Epigenetic regulation of varicella-zoster virus open reading frames 62 and 63 in latently infected human trigeminal ganglia [J]. J Virol, 2006, 80(10): 4921-4926.
[27] Mueller NH, Graf LL, Orlicky D, Gilden D, Cohrs RJ. Phosphorylation of the nuclear form of varicella-zoster virus immediate-early protein 63 by casein kinase II at serine 186 [J]. J Virol, 2009, 83(23): 12094-12100.
[28] Mueller NH, Walters MS, Marcus RA, Graf LL, Prenni J, Gilden D, Silverstein SJ, Cohrs RJ. Identification of phosphorylated residues on varicella-zoster virus immediate-early protein ORF63 [J]. J Gen Virol, 2010, 91(Pt 5): 1133-1137.
[29] Ambagala AP, Bosma T, Ali MA, Poustovoitov M, Chen JJ, Gershon MD, Adams PD, Cohen JI. Varicella-zoster virus immediate-early 63 protein interacts with human antisilencing function 1 protein and alters its ability to bind histones h3.1 and h3.3 [J]. J Virol, 2009, 83(1): 200-209.
[30] Ambagala AP, Cohen JI. Varicella-zoster virus IE63, a major viral latency protein, is required to inhibit the alpha interferon-induced antiviral response [J]. J Virol, 2007, 81(15): 7844-7851.
[31] Di Valentin E, Bontems S, Habran L, Jolois O, Markine-Goriaynoff N, Vanderplasschen A, Sadzot-Delvaux C, Piette J. Varicella-zoster virus IE63 protein represses the basal transcription machinery by disorganizing the pre-initiation complex [J]. Biol Chem, 2005, 386(3): 255-267.
[32] Johnson RW, Whitton TL. Management of herpes zoster (shingles) and postherpetic neuralgia [J]. Expert Opin Pharmacother, 2004, 5(3): 551-559.
[33] Connolly E, Braunstein S, Formenti S, Schneider RJ. Hypoxia inhibits protein synthesis through a 4E-BP1 and elongation factor 2 kinase pathway controlled by mTOR and uncoupled in breast cancer cells [J]. Mol Cell Biol, 2006, 26(10): 3955-3965.
[34] Du T, Zhou GY, Roizman B. Induction of apoptosis accelerates reactivation of latent HSV-1 in ganglionic organ cultures and replication in cell cultures [J]. Proc Natl Acad Sci USA, 2012, 109(36): 14616-14621.
[35] Workman A, Eudy J, Smith L, da Silva LF, Sinani D, Bricker H, Cook E, Doster A, Jones C. Cellular transcription factors induced in trigeminal ganglia during dexamethasone-induced reactivation from latency stimulate bovine herpesvirus 1 productive infection and certain viral promoters [J]. J Virol, 2012, 86(5): 2459-2473.
[36] Kim JY, Mandarino A, Chao MV, Mohr I, Wilson AC. Transient reversal of episome silencing precedes VP16-dependent transcription during reactivation of latent HSV-1 in neurons [J]. PLoS Pathog, 2012, 8(2): e1002540.
[37] Reichelt M, Zerboni L, Arvin AM. Mechanisms of varicella-zoster virus neuropathogenesis in human dorsal root ganglia [J]. J Virol, 2008, 82(8): 3971-3983.
[38] Zerboni L, Che XB, Reichelt M, Qiao YA, Arvin A. Herpes simplex virus 1 tropism for human sensory ganglion neurons in the severe combined immunodeficiency mouse model of neuropathogenesis [J]. J Virol, 2013, 87(5): 2791-2802.
[39] Esiri MM, Tomlinson AH. Herpes zoster:Demonstration of virus in trigeminal nerve and ganglion by immunofluorescence and electron microscopy [J]. J Neurol Sci, 1972, 15(1): 35-48.
. YAO Kun, E-mail: yaokun@njmu.edu.cn
Latent infection of varicella-zoster virus
TANG Huamin, JIA Junli, YAO Kun
DepartmentofImmunology,NanjingMedicalUniversity,Nanjing211166,China
Varicella-zoster virus (VZV) may cause latent infection and its reactivation is associated with zoster (shingles). The mechanism behind the VZV reactivation is still unclear. Here, the experimental models developed for VZV latency study are described and the updated knowledge is summarized.
Varicella-zoster virus; Latent infection; Animal model
国家自然科学基金(81273235、81571979)
姚堃
2016-06-01)
猜你喜欢
中成药(2021年5期)2021-07-21 08:39:04
广东医科大学学报(2020年6期)2020-02-06 06:00:46
中国中医急症(2019年10期)2019-05-21 07:20:40
山西教育·幼教(2017年2期)2017-04-23 06:56:51
现代检验医学杂志(2016年4期)2016-11-15 02:01:16
小学生时代·综合版(2016年5期)2016-05-14 17:53:48
灾害医学与救援(电子版)(2016年3期)2016-03-11 20:18:05
中国感染控制杂志(2015年7期)2015-12-13 08:30:42
发明与创新(2015年37期)2015-02-27 10:40:25
癌变·畸变·突变(2014年3期)2014-03-01 04:39:48
