
藤黄酸白蛋白纳米粒的制备及稳定性研究
2017-01-05 02:53杨志杰侯音璇
中国药剂学杂志(网络版) 2016年4期
杨志杰,侯音璇,林 霞,滕 欢,唐 星,徐 晖
(沈阳药科大学,药学院,辽宁,沈阳110016)
藤黄酸白蛋白纳米粒的制备及稳定性研究
杨志杰,侯音璇,林 霞,滕 欢,唐 星,徐 晖*
(沈阳药科大学,药学院,辽宁,沈阳110016)
目的以人血清白蛋白为载体制备藤黄酸白蛋白纳米粒,对处方和工艺进行优化,并进行稳定性考察。方法采用新型白蛋白纳米制备技术(NabTM)制备藤黄酸白蛋白纳米粒(GA-HSA NPs),用透射电镜观察纳米粒的形态,用Nicomp TM PSS 380进行粒度仪测定其粒度分布及电位,超速离心法测定其包封率、载药率及过膜效率,并考察其稀释稳定性及储存稳定性。结果GA-HSA NPs 电镜下呈球状粒子,粒径为(103.2 ± 34.4) nm, zeta 电位为-25.10 mV,GA-HSA NPs具有较高的包封率、载药率和过膜效率。GA-HSA NPs冻干粉针储存12个月期间无塌陷或皱缩,GA-HSA NPs混悬液在储存6周内粒径无显著性变化。结论难溶性抗癌药物藤黄酸可以采用NabTM制备成白蛋白纳米粒, 其粒径小, 稳定性高,有望成为藤黄酸的新型给药系统。
药剂学;纳米粒;新型白蛋白纳米制备技术;藤黄酸;稳定性;人血清白蛋白
藤黄酸(gambogic acid,GA)为藤黄科植物藤黄树(Garcinia hanbaryi Hook. f.)分泌的干燥树脂中提取纯化获得的有效成分[1]。近年研究发现,藤黄酸可显著抑制人肝癌及胃腺癌细胞的增殖,用于肿瘤治疗,具有活性成分性质稳定、疗效显著且不良反应少的特点,有望开发为一种高效低毒的抗肿瘤药物[2]。但是,藤黄酸在水中溶解度极小(小于0.5 mg·L-1),体内半衰期较短(狗体内小于1 h,兔子体内小于20 min),并且体内分布广泛,临床应用存在诸多困难[3]。DING Y等以聚氧乙烯蓖麻油和精氨酸用于尝试解决这些问题,但是这些溶剂可能会产生一系列的不良反应,如过敏反应、肾毒性、神经毒性和心脏毒性等[4]。因此,有必要开发一种新的剂型,以提高藤黄酸在水中的溶解度,减少溶剂带来的毒性及不良反应并提高其稳定性。
新型白蛋白纳米制备技术(nanoparticle-albumin bound technology,NabTM)是基于白蛋白带有巯基或二硫键基团,由高剪切力的气穴空化作用,生成使聚合物交联的超氧化物离子,氧化白蛋白中巯基残基或断裂白蛋白分子内/间现存的二硫键,使白蛋白内/间交联形成新的二硫键,从而在非水性介质微小液滴(如水不溶性药物)的周围形成一个交联的聚合物壳体。紫杉醇-白蛋白纳米混悬剂(nab-paclitaxel,Abraxane)已经获FDA批准上市,成为首个白蛋白纳米粒给药系统的成功案例。白蛋白可通过非共价键与物质紧密但可逆地结合,实现所运载物质在体内的运输和在细胞表面的释放,是疏水物质的体内天然载体。又由于白蛋白具有安全无毒、无免疫原性、可生物降解及生物相容性好等优点,是一种理想的靶向给药系统载体材料。因此,作者采用人血清白蛋白(human serum albumin,HSA)作为载体,利用溶剂蒸发技术,在没有任何聚合物核心材料和常规表面活性剂存在下成功制备藤黄酸白蛋白纳米混悬剂(GA-HSA NPs),并对其理化性质和稳定性进行考察。
1 仪器与材料
HITACHI 高效液相色谱仪(L-7420紫外检测器、L-7110泵、L-7200自动进样器)、HITACHI 紫外分光光度计、CS120GXL超高速离心机(日本日立公司),KQ-50B超声清洗机(昆山市超声仪器有限公司),高速分散匀质机(ULTRA TURRAX®T18 basic,IKA®WORKS,德国),PB-10酸度计(塞的利斯科学仪器有限公司),超声波细胞破碎机(美国SONICS公司),N1000旋转蒸发仪(东京理化器器械独资工厂),Nicomp TM PSS380 动态光散射粒度测定仪(Santa Barbara,USA PSS)。
藤黄酸原料药(江苏康缘药业有限公司),人血清白蛋白(兰州生物制品研究所有限公司),氯仿、二氯甲烷(分析纯,山东禹王实业有限公司),乙腈(色谱级,天津康科德科技有限公司),其他药品和试剂(药用规格和分析纯,市售)。
2 方法
2.1 GA-HSA NPs的制备(小试)
采用NabTM制备GA-HSA NPs。精密称取处方量的藤黄酸溶于有机相中(二氯甲烷与丙酮体积比4∶1),得含药油相。将人血清白蛋白加入注射用水中,搅拌使其分散均匀,用pH值为2的柠檬酸水溶液调节pH值为5.6~6.4,搅拌均匀得水相。将含药油相迅速加入水相中,然后转移至超声波细胞破碎机中,以300 W的功率超声乳化6 min (超声2 s,停1 s),即得O/W乳剂。将O/W乳剂在40 ℃条件下减压蒸发除去有机溶剂,用0.22 μm微孔滤膜过滤,冻干,即得注射用藤黄酸白蛋白纳米粒冻干品。
2.2 GA-HSA NPs的制备(放大至100 mL)
采用NabTM制备GA-HSA NPs。精密称取藤黄酸150 mg溶于有机相中(二氯甲烷与丙酮体积比4∶1),得含药有机相。将人血清白蛋白加至注射用水中,搅拌使其分散均匀,用pH值为2的柠檬酸水溶液调节pH值为5.6~6.4,搅拌均匀得水相。在高速分散机搅拌下,将含药油相缓慢加至水相中,待完全加入后以1×104r·min-1剪切3 min,即得初乳。将初乳转移至高压均质机中,以300 bar均质 3 次,500 bar均质3次,880 bar均质8次,即得O/W乳剂。将O/W乳剂在40 ℃条件下减压蒸发除去有机溶剂,用0.22 μm微孔滤膜过滤,冻干,即得注射用藤黄酸白蛋白纳米粒冻干品。
2.3 粒径及电位测定
使用NicompTM PSS 380进行粒度及电位测定。
2.4 样品含量测定
精密称取注射用藤黄酸白蛋白纳米粒冻干品适量,用10 mL蒸馏水复溶,即得GA-HSA NPs 含药溶液。精密量取上述含药溶液0.5 mL,置于 50 mL 量瓶中,用适量乙腈沉淀蛋白,水浴超声提取5 min,加乙腈稀释至刻度,摇匀,用0.22 μm微孔滤膜过滤。精密量取20 μL,注入色谱仪,记录色谱图,记录藤黄酸的峰面积。按外标法计算药物含量。
2.5 包封率、载药率及过膜效率的测定
本试验采用超速离心法对药物的包封率进行了测定。具体操作步骤如下。
2.5.1 药物总量的测定
精密称取注射用藤黄酸白蛋白纳米粒冻干品适量,用10 mL蒸馏水复溶,即得GA-HSA NPs 含药溶液。精密量取上述含药溶液0.5 mL,置于 50 mL 量瓶中,用适量乙腈沉淀蛋白,水浴超声提取5 min,加乙腈稀释至刻度,摇匀,用0.22 μm微孔滤膜过滤。用HPLC法测定制剂中藤黄酸的总浓度。
2.5.2 水相中药物含量的测定
取注射用藤黄酸白蛋白纳米粒溶液约 2.0 mL,置于超速离心管中,置离心机内以 4×104r·min-1转速离心约2 h,取离心管中的上清液体,用HPLC法测定水相中藤黄酸的浓度。
包封率计算公式如下:wEE= (mtotal– mfree) / mtotal×100%;载药率计算公式如下:wLC= min/mW×100%;过膜效率计算公式如下:wFE= mout/min×100%。
其中:mtotal为制剂中藤黄酸的总质量,mfree为水相中游离藤黄酸的总质量,载药率计算公式中的min为包裹在制剂中藤黄酸的总质量,mw为藤黄酸白蛋白纳米粒的总质量,mout为通过0.22 μm微孔滤膜过滤后藤黄酸的总质量,过膜效率计算公式中的min为未通过0.22 μm微孔滤膜过滤前藤黄酸的总质量。
3 结果与讨论
3.1 形态、粒径与电位的测定结果
藤黄酸白蛋白纳米粒的外观均为黄色均匀混悬液。裸眼观察无不溶性成分析出,显微镜下观察无藤黄酸不溶物析出。冻干产品为外观平整、色泽均匀、细腻疏松的块状物。使用 G220透射电镜进行纳米粒的形态观察(图1)。由图1可见,纳米粒粒径大小为100 nm左右,形态呈为圆整的球状,分布均匀,无粘连现象。使用Nicomp TM PSS 380进行粒度及电位测定(图2)。由图2可知,藤黄酸白蛋白纳米粒的粒径为(103.2 ± 34.4) nm,电位为-25.10 mV。

Fig. 1 Transmission electron microgram (TEM) image of GA-HSA NPs(× 35 000)图1 GA-HSA NPs 透射电镜照片(× 35 000)

Fig. 2 The particle size distribution (A) and the zeta potential (B) of GA-HSA NPs图 2 GA-HSA NPs的粒径分布(A)及zeta电位(B)
3.2 包封率、载药率及过膜效率
结果显示,藤黄酸白蛋白纳米粒的包封率、载药率和过膜效率分别为98.9%、10.9%和90.3%。
3.3 GA-HSA NPs的处方优化
3.3.1 有机相的影响
在应用NabTM制备白蛋白纳米粒时,不同种类的有机相对白蛋白纳米粒的形成以及稳定有重要影响。Rampon等研究发现,蛋白在界面上吸附后,蛋白的构象会发生变化,暴露出更多疏水集团,这些行为除了与蛋白质的性质有关外,还与非极性相的性质有关[5],因此,有机相的选择对于白蛋白纳米粒的制备至关重要。
本实验确定有机相与水相的体积比为1∶8,以细胞超声破碎仪为制备仪器,采用单因素考察法,设计有机相分别为二氯甲烷、乙酸乙酯、三氯甲烷、二氯甲烷-丙酮体积比2∶1和二氯甲烷-四氢呋喃体积比2∶1,考察不同种类的有机相组成及比例对藤黄酸白蛋白纳米的理化性质的影响,结果见图3。
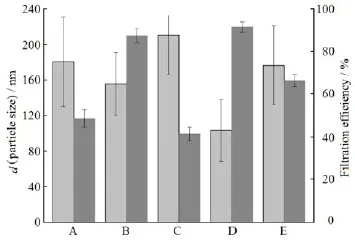
Fig. 3 Effect of the organic phase on the nanoparticle size and filtration efficiency图 3 有机相种类对GA-HSA NPs的粒度分布和过膜效率的影响
由图3 可知,混合有机相较单一有机相制备的纳米粒粒径分布更窄,且过膜效率更高。使用丙酮及四氢呋喃与二氯甲烷作为混合溶剂时,白蛋白纳米粒分布较窄、过膜效率高且有较好的稳定性。这可能与丙酮及四氢呋喃是与水互溶的溶剂有关,在制备过程中,丙酮和四氢呋喃能够适宜的破坏白蛋白水化膜,使白蛋白暴露更多的疏水基团以有利于药物包裹进去。综合考虑制剂稳定性及有机溶剂毒性的因素,最终确定有机相为二氯甲烷-丙酮体积比为2∶1。
3.3.2 相比的影响
在应用NabTM制备白蛋白纳米粒的过程中,相比的影响较为复杂。当有机相比下降时,有机相中的药物浓度增大,白蛋白不足以稳定所有新生界面,导致乳滴合并从而使粒径变大;当相比增大时,在除去有机溶剂过程中会有药物析出及乳滴合并。因此,选择合适的相比对于纳米粒的粒径有很大的影响。
本实验确定有机相为二氯甲烷-丙酮体积比 2∶1,人血清白蛋白的质量浓度为20 g·L-1。采用单因素考察法,设计有机相与水相的体积相比依次为1∶8、1∶10和1∶12,考察不同相比对于藤黄酸白蛋白纳米粒的理化性质的影响,结果见图4。

Fig. 4 Effect of the ratio of organic phase to water phase on the nanoparticle size and filtration efficiency (%)图4 相比对GA-HSA NPs的粒度分布及过膜效率的影响
由图4可知,当有机相与水相的体积相比为1∶8和1∶12时,纳米粒的粒径均大于150 nm,过膜效率均低于70%;当相比为1∶10时,纳米粒的粒径最小,过膜效率最高。
3.3.3 药物质量浓度的影响
考察不同药物质量浓度对藤黄酸白蛋白纳米粒粒度分布及过膜效率的影响。设计藤黄酸在有机相中的质量浓度分别为16、20、24、28和32 g·L-1,以确定最终藤黄酸白蛋白纳米粒的药物质量浓度范围,结果见表1。

Table 1 The influence of drug concentration on particle size distribution and filtration efficiency表1 不同药物质量浓度对GA-HSA NPs的粒度分布及过膜效率的影响
由表1中数据可知,药物质量浓度在16~28 g·L-1内,制备的白蛋白纳米粒粒度分布无明显变化,但是过膜效率呈下降趋势。当药物质量浓度达到32 g·L-1时,纳米粒子粒径显著增大、粒度分布变宽且过膜效率显著降低,说明有机相中药物质量浓度对纳米粒的粒度分布和过膜效率有显著影响。所以最佳药物质量浓度范围为24 ~ 28 g·L-1。
3.3.4 pH的影响
采用单因素考察法,设计不同白蛋白水溶液的pH值(pH值4.8 ~ 8.0),以确定最终藤黄酸白蛋白纳米粒的pH值范围,结果见图5。

Fig. 5 Influence of the pH of the HSA solution on the particle size and zeta potential图5 pH值对GA-HSA NPs的粒度分布及电位的影响
由图5可知,制备藤黄酸白蛋白纳米粒的最适pH值范围为5.6~6.4。在这个范围内所制备的藤黄酸白蛋白纳米粒粒径小、且电位稳定。当pH值小于5.6或者大于6.4时,药物的过膜效率较低,药物不能完全被包裹在纳米粒中。这可能与白蛋白的构象变化及药物白蛋白的相互作用有关,当pH值在5.6~6.4时,白蛋白呈现“N”构象,药物与白蛋白的亲和力较好,更有利于药物的包裹。而且在此范围内,藤黄酸(pKa = 7.8)主要以分子状态存在,推测藤黄酸与白蛋白的主要作用为疏水作用。
3.4 GA-HSA NPs的工艺优化(小试)
3.4.1 超声时间的影响
在应用NabTM制备白蛋白纳米粒的过程中,超声的主要作用是产生超氧化物离子交联蛋白以及为新生界面提供能量使粒径减小。作者采用单因素考察法,设计超声时间为1、3、6和9 min,考察不同超声时间对藤黄酸白蛋白纳米粒理化性质的影响,结果见图6。

Fig. 6 Effect of ultrasonic time on particle size distribution and filtration efficiency of GA-HSA NPs图6 超声时间对GA-HSA NPs的粒度分布及过膜效率的影响
由图6可知,随着超声时间的增加,纳米粒粒径逐渐减小,过膜效率逐渐增加。但是当超声时间超过6 min时,纳米粒粒径呈现增大趋势,过膜效率也随之降低。这可能是当超声时间小于6 min时产生的能量不足氧化白蛋白中巯基残基或断裂白蛋白分子内/间现存的二硫键, 从而不能使人血清白蛋白产生合适的构象以包裹药物。当超声时间大于6 min时,过量的能量输入会破坏已经产生的稳定体系,使纳米粒系统出现超负荷(overprocessing)现象,造成纳米粒粒径增大,过膜效率降低。
3.4.2 超声功率的影响
采用单因素考察法,设计超声功率为200、300、400和500 W,考察不同超声功率对藤黄酸白蛋白纳米粒理化性质的影响,结果见图7。

Fig. 7 Effect of ultrasonic power on particle size distribution and filtration efficiency of GA-HSA NPs图7 超声功率对GA-HSA NPs的粒度分布及过膜效率的影响
由图7可知,当超声功率由200 W增加到300 W时,纳米粒粒径显著降低,而当超声功率逐渐增加到500 W时,粒径却有上升趋势。由此推测,当超声功率由200 W增加到300 W时,白蛋白的交联程度、疏水性及所带电荷所形成的交联聚合物壳体,正适合包裹藤黄酸类型药物。
3.4.3 减压除有机溶剂温度的影响
分别在25、30、40、50和60 ℃条件下减压除去有机溶剂。考察减压除去有机溶剂的温度对藤黄酸白蛋白纳米粒理化性质的影响,结果见图8。
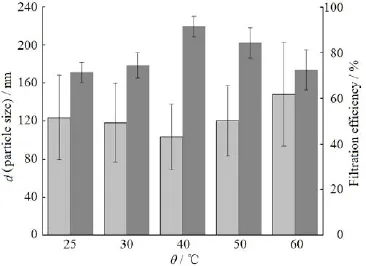
Fig. 8 Effect of evaporation temperature of organic solvent on particle size distribution and filtration efficiency of GA-HSA NPs图8 减压除有机溶剂温度对GA-HSA NPs的粒度分布及过膜效率的影响
由图8可知,减压除去有机溶剂的温度为40 ℃时,纳米粒的粒径为100 nm左右,过膜效率大于90%。温度高于或低于40 ℃时,纳米粒的粒径增大,过膜效率降低。
3.5 GA-HSA NPs的工艺优化(放大至100 mL)
3.5.1 高速剪切机剪切速度和剪切时间影响
作者对采用高速剪切制备初乳的制备条件进行了考察,对剪切速度和剪切时间进行了优化。
以初乳的外观和显微镜下乳滴的均匀度来评价高速剪切的条件。分别以 6 000、8 000、1×104和1.2×104r·min-1的剪切转速,剪切 3 min 制备初乳,在显微镜下观察乳滴的均匀度。结果表明:当高速剪切的转速为 1×104r·min-1时,可得粒度分布均匀的乳滴;当剪切速度超过最佳条件后,由于剪切力的作用,使乳滴的碰撞几率增加,剪切均匀的乳滴进一步合并增大。
固定剪切转速为 1×104r·min-1,考察剪切时间分别为 1、3和5 min 时,剪切时间对初乳的影响。结果,乳化时间为 1 min 时,乳滴粒度分布不均匀;乳化时间为 3 min 和 5 min 时,对初乳的外观影响较小,均为黄色混悬液,在显微镜下观察可知粒径也无显著差别。由于高速剪切功率有限,只能将初乳粒径减小到一定程度,之后即使再延长剪切时间,粒径也不会减少,反而会产生一定热量。因此最终确定高速剪切的条件为1×104r·min-1高速剪切 3 min。
3.5.2 高压均质过程的影响
高压均质过程对纳米粒的粒径和粒径分布有很大影响,因此,本实验对高压均质操作参数进行了优化。本实验先以300 bar和500 bar低压匀化经剪切所制得的初乳,然后经800 bar高压过程来减小乳滴粒径。着重考察高压(800 bar)次数对粒径及粒径分布的影响,结果见表2。

Table 2 The influence of homogenization cycles on the characteristics of GA-HSA NPs (n=3)表2 高压均质次数对GA-HAS NPs的粒度分布的影响 ( n=3)
由表2可知,当高压均质次数由3次升到8次的时候,平均粒径不断减小,分布不断变窄;当高压均质次数在8以上时,随均质次数的增加,平均粒径虽然变化不大,但是粒度分布逐渐变宽。这可能是由于过多的均质次数导致乳滴两级分化,乳滴分布不均,在一定程度上增加了乳滴的相互碰撞,引起乳滴的合并。综合上述试验结果,最终确定制备藤黄酸白蛋白纳米粒的高压(800 bar)均质次数为8次。
3.6 冻干保护剂的筛选
将“2.1”条下制备的GA-HAS NPs混悬液分别加入100 g·L-1甘露醇、100 g·L-1乳糖和100 g·L-1蔗糖,摇匀后按照如下工艺进行冷冻干燥(图9)。以GA-HSA NPs冻干后的外观、水化难易程度及复溶后的粒径、过膜效率等为评价指标,结果见表3。

Fig. 9 The lyophilization curve of GA-HSA NPs图9 GA-HAS NPs的冻干曲线

Table 3 Characteristics of lyophilized GA-HSA NPs using different lyoprotectants表3 不同冻干保护剂制备得到的GA-HSA NPs混悬剂的冻干粉针
由表3可知,不添加任何冻干保护剂制得的GA-HAS NPs冻干粉针与添加各种冻干保护剂的形态、外观、水化难易程度差异不大,并且不添加冻干保护剂复溶前后粒径变化不大。故本实验GA-HAS NPs的冻干制备过程中可不添加冻干保护剂。这是由于体系中的HSA既作为载体又可作为冻干保护剂,使得到的冻干产品不仅具有良好的外观而且具有长期稳定性。
3.7 稳定性考察
3.7.1 稀释稳定性考察
纳米粒在不同条件下的稳定性是影响药物制剂应用的重要因素。当药物进入体内以后,血液成分可能会对载药纳米粒的稳定性产生影响,因此需要进行纳米粒的血清稳定性实验。一般来说血液体积是人体体质量的7%左右,而当静脉给药时局部血液体积是未知的,所以将制剂稀释不同倍数以考察其稀释稳定性。
稀释介质:生理盐水作为等渗溶液的稀释介质;用人血清白蛋白模仿体内蛋白环境,用磷酸盐缓冲液(pH值7.4 PBS)维持细胞渗透压,调节培养液酸碱平衡,故选用含50 g·L-1人血清白蛋白的PBS作为血清稳定性的稀释介质。
将制备好的GA-HSA NPs冻干粉分别用等渗溶液(生理盐水)和50 g·L-1人血清白蛋白的PBS复溶并稀释,稀释倍数依次为2、8、32和100倍。通过比较稀释前后粒径及粒度分布(P.I.)来考察不同稀释倍数的稳定性,结果见图10。
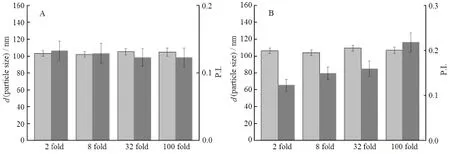
Fig. 10 The effect of different x-fold dilutions on GA-HSA NPs图10 不同稀释倍数对GA-HAS NPs的影响
由图10A可知,GA-HSA NPs冻干粉用等渗溶液(生理盐水)复溶并稀释不同倍数后,粒径及粒度分布(P.I.)无显著性增加或减小,且稀释过程中无沉淀产生。这说明GA-HSA NPs经等渗溶液(生理盐水)稀释不同倍数后,具有很强的稳定性。
由图10B可知,GA-HSA NPs经50 g·L-1人血清白蛋白稀释后,在稀释倍数由2倍增加到8倍时,纳米粒的粒径无显著性变化,而粒度分布(P.I.)却逐渐增加。这可能是由于内源性蛋白质吸附到纳米粒上,使其粒径分布(P.I.)增加,但是这种增加在可接受的范围内。这说明GA-HSA NPs在模拟体内蛋白环境下具有很好的稳定性。
3.7.2 储存稳定性考察
为考察GA-HSA NPs长期储存稳定性。根据国际协调会议的要求,将GA-HSA NPs的冻干粉和混悬液分别放置在4 ℃及25 ℃、相对湿度为60%的条件下。通过测定储存期间纳米粒的粒径及粒度分布的变化,考察其在以上两种条件下的稳定性,结果见图11。
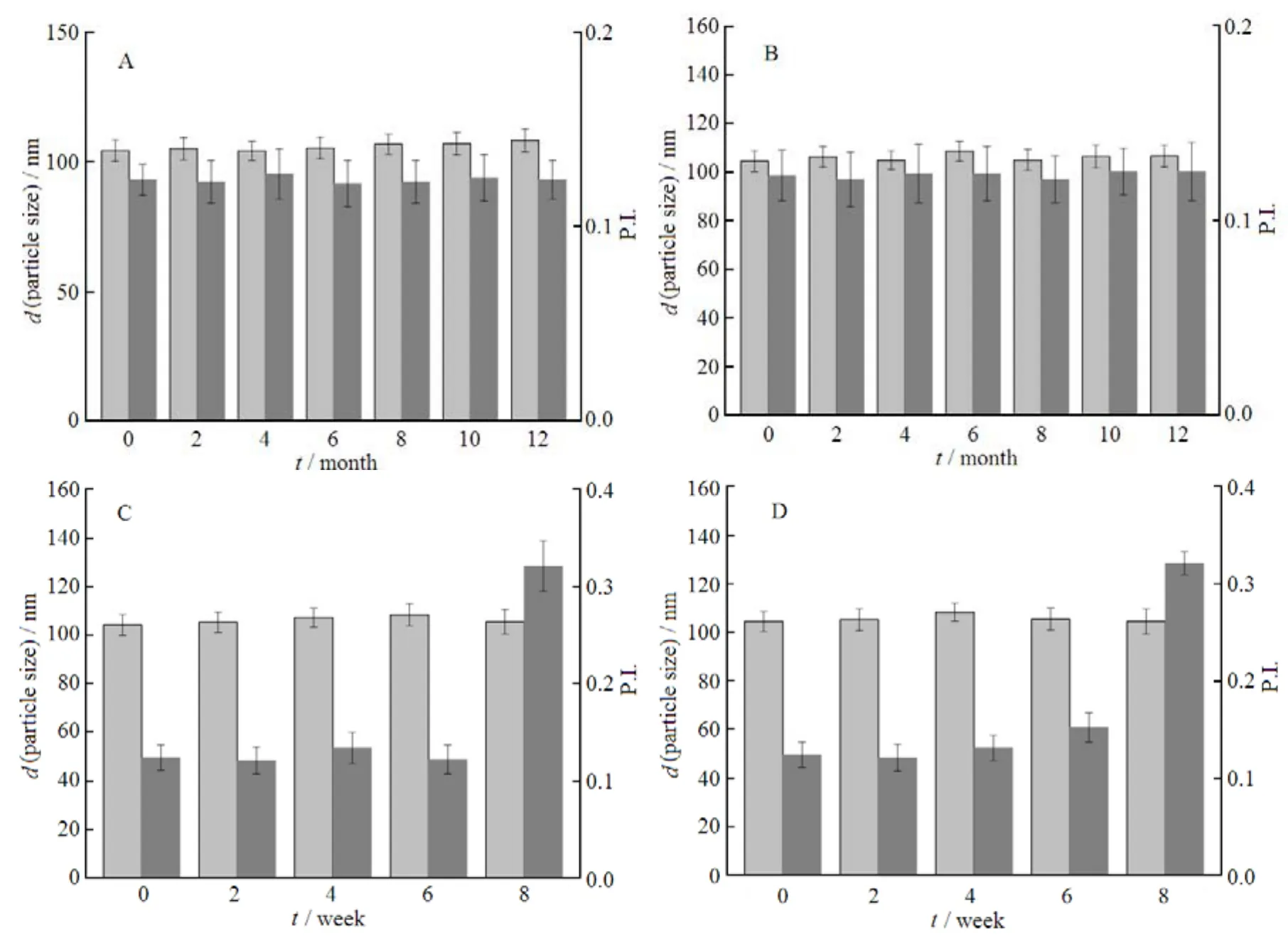
Fig. 11 The stability of GA-HSA NPs图11 GA-HAS NPs储存稳定性考察
由图11A及11B可知,在储存12个月期间,GA-HSA NPs的冻干粉在4 ℃及25 ℃、相对湿度60%条件下,纳米粒的粒径及粒度分布无显著性增大或者减小,且冻干粉外观上色泽均匀、无皱缩或塌陷,储存稳定性良好。由此说明,冻干储存方式可以作为藤黄酸白蛋白纳米粒的一种有效的、长期储存方法。
由图11C及11D可知,GA-HSA NPs的混悬液在4 ℃及25 ℃、相对湿度60%条件下,纳米粒的粒径及粒度分布无显著性增大或者减小,但是在储存6周之后,纳米粒的分布显著变大,且两种条件下外观上观察到有药物析出。引起这种现象的原因可能是在长期的储存过程中,小粒子不断聚集成大粒子造成大粒子的药物浓度增大,这样在大粒子与小粒子之间形成浓度梯度。当大粒子吸附的药物饱和时即会有药物沉淀出来。所以在临床使用时,GA-HSA NPs的混悬液存放日期应在6周以内,但在实际使用时一般是现配现用,所以GA-HSA NPs的混悬液在6周内稳定为临床使用提供了有利保障。
4 结论
难溶性抗癌药物藤黄酸可以采用NabTM制备成白蛋白纳米粒, 其粒径小,分布均匀,稳定性好,为临床使用提供了有利保障。且操作简单,可放大,有望成为藤黄酸的新型给药系统。
[1] 侯文洁,萧伟. 藤黄酸的研究进展[J]. 中草药, 2011, 42(3): 617-620.
[2] 王鸣,冯煦,赵友谊, 等. 中药藤黄的研究和应用[J]. 中国野生植物资源, 2003, 22 (1): 1-4.
[3] HAO K, LIU X Q, WANG G J, et al. Pharmacokinetics, tissue distribution and excretion of gambogic acid in rats[J]. Eur J Drug Metab Pharmacokinet, 2007, 32, 63-68.
[4] DING Y, ZHANG P, TANG X Y, et al. PEG prodrug of gambogic acid: Amino acid and dipeptide spacer effects[J]. Polymer, 2012, 53, 1694-1702.
[5] RAMPON V, BROSSARD C, MOUHOUS-RIOU N, et al. The nature of the apolar phase influences the structure of the protein emulsifier in oil-in-water emulsions stabilized by bovine serum albumin: A front-surface fluorescence study[J]. Adv Colloid Interfac, 2004, 108: 87-94.
Studies on preparation and stability of human serum albumin nanoparticles containing gambogic acid
YANG Zhijie, HOU Yinxuan, LIN Xia, TENG Huan, TANG Xing, XU Hui*
(School of Pharmacy, Shenyang Pharmaceutical University, Shenyang 110016, China)
ObjectiveTo develop human serum albumin (HSA) nanoparticles (NPs) as polymeric vehicles for intravenous injection of gambogic acid (GA), to optimize the formulation thereof and evaluate its stability.MethodsGA-HSA NPs were prepared by the nanoparticle albumin-bound technology (Nab™). The morphology of the NPs was observed by TEM. The distribution of particle size and zeta potential of the NPs were determined by NicompTM PSS 380. The encapsulation efficiency (EE), drug-loading capacity (GA-LC) and filtration efficiency of the NPs were examined by ultracentrifugation method. Andthe stability upon dilution and storage stability were also investigated.ResultsGA-HSA NPs showed spherical in shape with a particle size of (103.2 ± 34.4) nm and a zeta potential of -25.10 mV. And high entrapment efficiency was achieved. Freeze–dried GA-HSA NPs showed no collapse or shrinkage after 12 months, and GA-HSA NP suspension demonstrated significant changes within 6 weeks.ConclusionsThis formulation could significantly increase the concentration of GA in water. GA-HSA NPs prepared by this method exhibited a small particle size, high stability. Therefore, this delivery system is a promising polymeric carrier for the transport of GA.
pharmaceutics; nanoparticle albumin-bound technology (NabTM); nanoparticles; gambogic acid; stability;human serum albumin
R 94
A
(2016)04–0107–13
10.14146/j.cnki.cjp.2016.04.001
(本篇责任编辑:赵桂芝)
2015–04–29
杨志杰(1989–),女(汉族),内蒙古赤峰人,硕士研究生,E-mailyangzhijie26@126.com; *
:徐晖(1972–), 男(汉族), 辽宁法库人, 副教授, 主要从事药剂学研究,Tel.024–23986356,E-mailxuhui-spu@163.com。
猜你喜欢
中国饲料(2021年17期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
粉末冶金技术(2021年3期)2021-07-28
世界最新医学信息文摘(2021年12期)2021-06-09
现代临床医学(2021年2期)2021-03-29
河南畜牧兽医(2021年10期)2021-01-05
小型微型计算机系统(2020年10期)2020-10-21
中成药(2018年4期)2018-04-26
数码设计(2017年1期)2017-10-13
中南大学学报(自然科学版)(2015年9期)2015-10-13

- 中国药剂学杂志(网络版)的其它文章
- 帕洛诺司琼贴剂的含量测定方法
- 纳豆激酶与尿激酶的对比性研究
- 蚕丝蛋白对磷酸钙颗粒释药的影响