
常染色体隐性遗传性耳聋*
2016-12-24 06:06:30王秋菊袁永一
听力学及言语疾病杂志 2016年4期
王秋菊袁永一
·继续教育园地·
常染色体隐性遗传性耳聋*
王秋菊1袁永一1
常染色体隐性遗传性耳聋是指与耳聋表型相关的基因位于常染色体上,此类耳聋只有在两个分别来自父母的等位基因均携带致病突变时才出现耳聋。有关常染色体隐性遗传性耳聋的描述最早出现在16世纪,Schenck描述了一个家系中的多名兄弟姐妹均患有先天性重度耳聋,而他们的父母听力完全正常。1853年,Wilde提出在常染色体隐性遗传性耳聋中前瞻性调查父母血亲关系十分重要。但在1875年George Darwin否定了这一观点。1877年,一项匿名研究指出,在Marsha's Vineyard,所有后代均来自共同的祖先,其中60%患有耳聋。1880年Hartmann提供了耳聋显性及隐性遗传的证据,他也强调在隐性遗传性耳聋中研究父母近亲婚配的重要性。
常染色体隐性遗传性耳聋具有以下特征:①致聋基因位于常染色体,因而致聋基因的遗传与性别无关,即男、女性的患病机会均等;②系谱中通常看不到连续传递现象,往往是散发病例,但同胞中可有多人患病;③患者的双亲一般不患病,但都是致病基因的携带者;④患者的同胞有1/4的风险患病,患者表型正常的同胞中有2/3的概率为携带者;⑤患者的后代一般不发病,但一定是携带者;⑥近亲婚配时子女的发病风险显著提高,因为共同的祖先可能传递给他们共同的突变基因。
一般所指的常染色体隐性遗传性耳聋多指非综合征型耳聋(autosomal recessive nonsyndromic hearing loss,ARNSHL),耳聋是其唯一临床表现,大多数患者症状早发且程度较重。部分综合征型耳聋也遵循常染色体隐性遗传模式,除了耳聋表型外同时伴有其他器官系统功能异常,比如Pendred综合征伴有甲状腺肿,Usher综合征伴有渐进性视网膜色素变性(多为儿童期末至青春期发病)而致的视野缩小、视力障碍等。
1 常染色体隐性遗传性耳聋致病基因的命名原则和基因的发现
常染色体隐性遗传性耳聋,英文表述为autosomal recessive hereditary hearing loss,缩略词为ARHHL。下面对常染色体隐性遗传性耳聋致病基因的座位及致病基因的发现进行简述。
1.1 致病基因的座位 根据人类基因组命名委员会的规则,以DFNB来表示常染色体隐性遗传性耳聋,DFN缩略词来自deafness中的三个字母,B定义为隐性遗传。第一个致病基因座位表述为DFNB1,此后依据基因座发现及获得命名的时间顺序,依次命名为DFNB2,DFNB3……目前,常染色体隐性遗传的基因座位一共有86个,命名从DFNB1到DFNB103,其中空缺的座位包括DFNB34,41,43,50,52,54,56,57,58,64,69,70,75,78,87,92,100。
1.2 致病基因的发现 常染色体隐性遗传性耳聋中致病基因明确的基因座位达56个,迄今致病基因未明的有30个。DFNB1和DFNB18座位上有两个致病基因,其他基因座分别对应一个致病基因。DFNB7和DFNB11,DFNB8和DFNB10、DFNB15和DFNB72及DFNB95、DFNB66和DFNB67分别被证实具有相同的致病基因。
提及常染色体隐性遗传性耳聋致病基因的发现,不得不提到GJB2基因,它是目前各人种最主要的致聋因素之一。1997年,学者们通过连锁分析候选基因筛查的方法,将DFNB1座位的致病基因确定为编码缝隙连接蛋白(connexin26,Cx26)的GJB2,该基因突变还能导致常染色体显性遗传性聋DFNA3(OMIM601544)。GJB2基因于1993年被克隆,定位于染色体13q11-12。随着研究的深入,人们发现在一些定位于DFNB1座位的耳聋家系中未发现GJB2突变,因而考虑这一区域还存在其他致聋基因,这就是位于GJB2端粒端上游与其相隔约35 kb的GJB6。2002年研究人员鉴定了另一个致聋基因GJB6,该基因于1999年被克隆,位于染色体13q12。为了将GJB2和GJB6所对应的基因座位精确区分,又将DFNB1座位细分为DFNB1A和DFNB1B。在常染色体隐性遗传性耳聋中,类似同一基因座位有两个致病基因的座位还包括DFNB18,其致病基因包括USH 1C和OTOG,该两个基因分别位于染色体11p14.3和11p15.1。
2 常染色体隐性遗传性耳聋的表型特点
ARNSHL临床表现多数为双耳语前中、重度-极重度感音神经性耳聋,但少数耳聋基因如GJB2、SLC26A4、TECTA、TMPRSS3、MYO3A等可表现为语后迟发性、中度感音神经性耳聋,甚至某些频率的听力出生时可在正常范围内。各个常染色体隐性遗传性耳聋基因对应的表型见表1。

表1 常染色体隐性遗传性NSHL的基因、定位、表达及表型
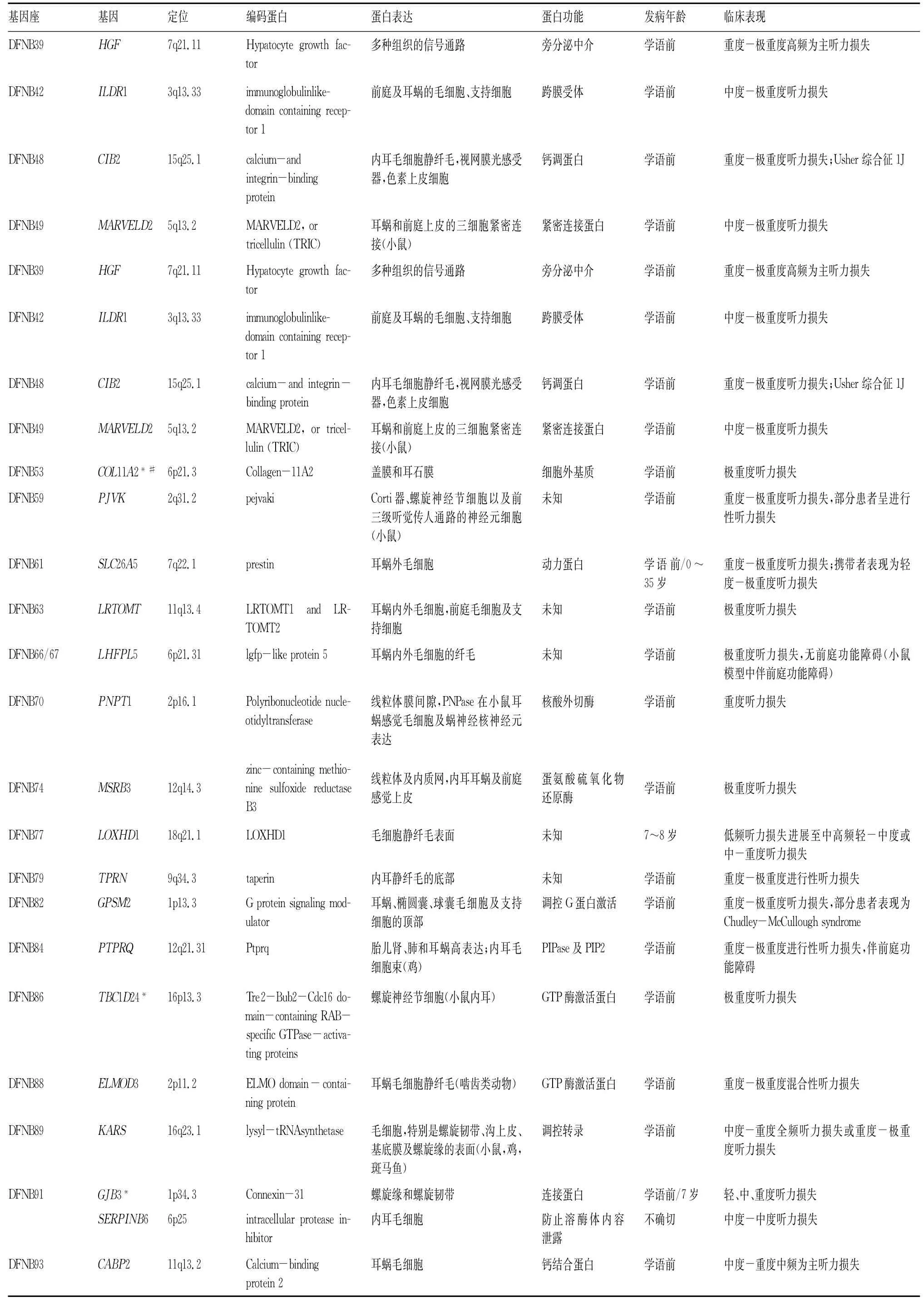
蛋白功能发病年龄临床表现旁分泌中介学语前重度-极重度高频为主听力损失跨膜受体学语前中度-极重度听力损失钙调蛋白学语前重度-极重度听力损失;Usher综合征1J紧密连接蛋白学语前中度-极重度听力损失tor 1旁分泌中介学语前重度-极重度高频为主听力损失跨膜受体学语前中度-极重度听力损失DFNB48 CIB2 15q25.1 calcium-and integrinbinding protein DFNB49 MARVELD2 5q13.2 MARVELD2,or tricellulin(TRIC)内耳毛细胞静纤毛,视网膜光感受器,色素上皮细胞耳蜗和前庭上皮的三细胞紧密连接(小鼠)钙调蛋白学语前重度-极重度听力损失;Usher综合征1J紧密连接蛋白学语前中度-极重度听力损失DFNB53 COL11A2*#6p21.3 Collagen-11A2盖膜和耳石膜细胞外基质学语前极重度听力损失DFNB59 PJVK 2q31.2 pejvaki Corti器、螺旋神经节细胞以及前三级听觉传人通路的神经元细胞(小鼠)未知学语前重度-极重度听力损失,部分患者呈进行性听力损失DFNB61 SLC26A5 7q22.1 prestin耳蜗外毛细胞动力蛋白学语前/0~35岁重度-极重度听力损失;携带者表现为轻度-极重度听力损失DFNB63 LRTOMT 11q13.4 LRTOMT1 and LRTOMT2耳蜗内外毛细胞,前庭毛细胞及支持细胞未知学语前极重度听力损失DFNB66/67 LHFPL5 6p21.31 lgfp-like protein5耳蜗内外毛细胞的纤毛未知学语前极重度听力损失,无前庭功能障碍(小鼠模型中伴前庭功能障碍)DFNB70 PNPT1 2p16.1 Polyribonucleotide nucleotidyltransferase线粒体膜间隙,PNPase在小鼠耳蜗感觉毛细胞及蜗神经核神经元表达核酸外切酶学语前重度听力损失DFNB74 MSRB3 12q14.3 zinc-containing methionine sulfoxide reductase B3线粒体及内质网,内耳耳蜗及前庭感觉上皮蛋氨酸硫氧化物还原酶学语前极重度听力损失DFNB77 LOXHD1 18q21.1 LOXHD1毛细胞静纤毛表面未知7~8岁低频听力损失进展至中高频轻-中度或中-重度听力损失DFNB79 TPRN 9q34.3 taperin内耳静纤毛的底部未知学语前重度-极重度进行性听力损失DFNB82 GPSM2 1p13.3 Gprotein signaling modulator调控G蛋白激活学语前重度-极重度听力损失,部分患者表现为Chudley-McCullough syndrome DFNB84 PTPRQ 12q21.31 Ptprq胎儿肾、肺和耳蜗高表达;内耳毛细胞束(鸡)耳蜗、椭圆囊、球囊毛细胞及支持细胞的顶部PIPase及PIP2学语前重度-极重度进行性听力损失,伴前庭功能障碍DFNB86 TBC1D24*16p13.3 Tre2-Bub2-Cdc16 domain-containing RAB-specific GTPase-activating proteins DFNB88 ELMOD3 2p11.2 ELMO domain-containing protein螺旋神经节细胞(小鼠内耳)GTP酶激活蛋白学语前极重度听力损失耳蜗毛细胞静纤毛(啮齿类动物)GTP酶激活蛋白学语前重度-极重度混合性听力损失DFNB89 KARS 16q23.1 lysyl-tRNAsynthetase毛细胞,特别是螺旋韧带、沟上皮、基底膜及螺旋缘的表面(小鼠,鸡,斑马鱼)调控转录学语前中度-重度全频听力损失或重度-极重度听力损失DFNB91 GJB3*1p34.3 Connexin-31螺旋缘和螺旋韧带连接蛋白学语前/7岁轻、中、重度听力损失SERPINB6 6p25 intracellular protease inhibitor内耳毛细胞防止溶酶体内容泄露不确切中度-中度听力损失DFNB93 CABP2 11q13.2 Calcium-binding protein2耳蜗毛细胞钙结合蛋白学语前中度-重度中频为主听力损失

注:学语前听力损失包括先天性听力损失,DFNB8性听力损失的发病在学语后(10~12岁),DFNB10型听力损失发病为先天性,*标记的基因既可引起隐性遗传性听力损失也可引起显性遗传性听力损失,共9个,#标记的基因既可引起隐性遗传性听力损失也可引起耳聋综合征,共6个
3 常染色体隐性遗传性耳聋的遗传异质性(genetic heterozygenity)
ARNSHL常见的遗传异质性包括两种类型:等位型(allelic)与非等位型(non-allelic)。等位型的遗传异质性是指同一基因的不同突变在不同情况下产生同样的耳聋表型;非等位型的遗传异质性则是指不同基因的突变导致相同或相似的耳聋表型。在家系研究中非等位型的异质性可以通过家系的连锁分析(linkage analysis)区分开,而等位型的异质性则难以区分。例如,导致家族性耳聋的突变基因超过100个,针对某个家系,其致病突变可能发生在某个单一的基因上,因此,不同的家系其致病突变就可能发生在不同的基因上,这种非等位型的异质性在采用连锁分析对不同的家系进行分析时就可以得到不同的连锁位点。等位型的遗传异质性通过连锁分析一般不能区分,只有对基因进行详细的测序后才可以得出结果。在常染色体隐性遗传性耳聋的致聋基因中,有一些除了与DFNB有关,还与DFNA有关,如GJB 2、GJB 6、GJB 3、TECTA、MYO7A、MYO6、COL 11A 2、TMC1和TBC1D 24。此外,一些DFNB基因还能导致综合征型耳聋,如GJB 2(DFNB1A)突变导致耳聋-皮肤掌跖角化综合征和角膜炎-鱼鳞病-耳聋综合征(keratitis-ichthyosis-deafness syndrome,KID),SLC26A4(DFNB4)突变导致Pendred综合征,MYO7A(DFNB2)能导致Usher 1B,CDH 23(DFNB12)能导致Usher 1D, PCDH 15(DFNB23)能导致Usher 1F,COL 11A 2(DFNB53)能导致Stickler综合征。
4 常染色体隐性遗传性耳聋的婚配类型及子代发病风险的预测
对于常染色体隐性遗传性耳聋,突变基因为等位基因a,呈隐性,只有基因型为aa纯合子时才表现为疾病,纯合子AA或杂合子Aa表型正常。两个致病基因分别来自患者双亲,因而患者双亲都是携带一个致病基因的杂合子Aa;虽然表型正常,但再次生育时仍可能把致病基因传给后代。两个常染色体隐性遗传病的肯定携带者(obligate carrier)婚配后,其子女的发病风险为1/4;若其子女表型正常,则有2/3的概率是携带者。
1 Toriello HV,Reardon W,Gorlin RJ.Hereditary hearing loss and its syndromes(Second edition).Oxford University Press,2004.1~2.
2 http://hereditaryhearingloss.org/Hereditary Hearing loss Homepage.
3 http://www.omim.org/.
4 王秋菊,韩东一,翟所强m等.遗传性听力损失及其综合征[M].第2版.北京:人民军医出版社,2006.79~103.
5 Toriello HV,Smith SD.Hereditary hearing loss and its syndromes(Third edition)[M].Oxford University Press,2013.147~200.
(2016-06-22收稿)
(本文编辑 周涛)
10.3969/j.issn.1006-7299.2016.04.026
R764.44
A
1006-7299(2016)04-0421-04
* 国家重大科学研究计划项目(2014CB943001)、国家自然基金重点国际合作项目(81120108009)、国家自然科学基金重点项目(81530032)联合资助
1 解放军总医院耳鼻咽喉头颈外科,解放军耳鼻咽喉研究所(北京100853)
王秋菊(Email:wqcr@263.net或wqcr301@sina.com)
猜你喜欢
中国现代医生(2022年19期)2022-11-04 10:13:29
昆明医科大学学报(2022年4期)2022-05-23 13:04:50
中国临床医学影像杂志(2021年10期)2021-11-22 07:46:36
基层中医药(2021年8期)2021-11-02 06:24:54
中国民间疗法(2021年8期)2021-07-22 05:53:42
中国生殖健康(2020年4期)2021-01-18 02:58:32
基础医学与临床(2020年6期)2020-02-12 12:03:38
中华耳科学杂志(2020年6期)2020-01-08 07:40:33
中国生殖健康(2018年4期)2018-11-06 07:12:36
中华耳科学杂志(2018年6期)2018-01-16 13:45:59
