
阿托伐他汀对大鼠脑缺血再灌注PERK/elfR2a通路及Caspase-3表达的影响
2016-12-24 19:31,,,
中南医学科学杂志 2016年1期
,, ,
(南华大学附属省马王堆医院神经内科,湖南 长沙 410015)
·基础医学·
阿托伐他汀对大鼠脑缺血再灌注PERK/elfR2a通路及Caspase-3表达的影响
彭文娟,杨剑文*,刘湘玉,杨期明
(南华大学附属省马王堆医院神经内科,湖南 长沙 410015)
目的研究蛋白激酶R样内质网激酶(PERK)/eIFR2a通路及Caspase-3在大鼠脑缺血再灌注损伤中的作用机制及阿托伐他汀对其的影响。方法采用大脑中动脉线栓塞法制作大鼠脑缺血再灌注模型;随机分为缺血再灌注组、假手术组、阿托伐他汀组、阿托伐他汀+Salubrinal抑制剂组,大体标本采用TTC染色,釆用Western-blot法检测PERK、Caspase-3蛋白表达及eIF2a蛋白磷酸化。结果与假手术组相比,大鼠缺血再灌注后PERK蛋白表达及eIF2a 的磷酸化增加, Caspase-3表达的活性增强(P<0.01);阿托伐他汀干预可以减轻PERK蛋白表达及eIF2a蛋白磷酸化(P<0.05)。给予特异性eIF2a磷酸化抑制剂Salubrinal后可抑制eIF2a的磷酸化及Caspase-3表达的活性(P<0.05),对PERK蛋白表达无影响。形态学上从TTC染色提示:在缺血再灌注组TTC染色可见大片脑梗死组织。Salubrinal抑制剂及阿托伐他汀干预后脑梗死体积明显缩小(P<0.05)。结论内质网应激通过PERK/eIF2a/Caspase-3途径促进细胞凋亡,阿托伐他汀干预可以减轻脑缺血再灌注损伤。
蛋白激酶R样内质网激酶; eIF2a; 内质网应激; Caspase-3; 局灶性脑缺血再灌注; 阿托伐他汀
脑梗死发生后出现脑细胞的坏死,神经损伤加重,其损伤的机制主要与细胞凋亡有关,最近几年有大量研究发现内质网应激(ERS)与细胞凋亡有关,内质网是一种细胞器,参与了蛋白质的折叠、稳态和脂质合成等过程,多种诱导因素包括氧化应激、缺血、电解质紊乱等影响内质网功能,导致非折叠蛋白和错误折叠蛋白在内质网的积聚,触发内质网应激,诱导促凋亡转录因子C/EBP同源蛋白(C/EBP homologousprotein,CHOP),激活C-JNK激酶、分裂凋亡蛋白12及/Caspase-3等,从而启动细胞凋亡或死亡,启动细胞凋亡信号[1-4]。
内质网应激包含蛋白激酶R样内质网激酶(protein kinase R-like ER kinase,PERK)、肌醇激酶1(inositol-requiring kinase 1,IRE-l)和转录激活因子6(transcriptional factor activating transcription factor 6,ATF-6)3条信号通路[5-9]。其中PERK是一种丝氨酸苏氧酸激酶,在内质网应激启动后被激活,还能磷酸化翻译起始因子2a(eIF2a),从而切断mRNA的翻译过程。Caspase-3是引起细胞凋亡的关键酶之一,是执行凋亡程序的下游关键因子之一,因而推测缺血再灌注等启动内质网应激导致细胞凋亡可能与Caspase-3有关,且PERK/eIF2a/Caspase-3通路是其导致脑组织损伤机制之一。
近期有研究报道,氟伐他汀可以抑制缺氧所导致的巨噬细胞凋亡,其机制与抑制内质网应激有关,参与内皮细胞的凋亡[10-11]。而阿托伐他汀与氟伐他汀具有类似的分子结构,能竞争性抑制羟甲基戊二酰辅酶A还原酶(HMG-CoA),因此推测阿托伐他汀也可能通过内质网应激途径抑制缺氧等所导致细胞凋亡,减轻脑缺血再灌注损伤,从而起到神经保护作用。本研究拟探索阿托伐他汀对缺血再灌注损伤内质网应激的作用机制。
1 材料与方法
1.1脑缺血再灌注动物模型的建立清洁级健康雄性SD大鼠60只,鼠龄10个月,体质量89±9 g,购自中南大学医学院动物实验室(实验动物许可证号SYXK(湘)2013-001)。大鼠的饲养温度20~25℃,湿度40%~70%,室内风速0.1~0.2 m/s,光照时间为12 h/12 h明暗。实验经本院动物伦理委员会批准同意。按数字随机法将大鼠分为4组:假手术组、缺血再灌注组、阿托伐他汀组和阿托伐他汀+Salubrinal抑制剂组(简称阿+抑制剂组),每组15只。缺血再灌注组、阿托伐他汀组和阿+抑制剂组大鼠以大脑中动脉闭塞法建立脑缺血再灌注模型,用10%水合氯醛350 mg/kg麻醉。切开颈前正中皮肤,钝性分离皮下组织,暴露右侧颈总动脉及颈内外动脉,在颈总动脉距离颈动脉分叉2 cm处剪一斜口,将标记好的栓线通过分叉插向颈内动脉,插入深度为18.5±1.0 mm,表明线栓已经堵塞大脑中动脉血流,栓塞2 h后再灌注恢复血流,最后缝合皮肤。假手术组大鼠暴露右侧颈总动脉。其中阿托伐他汀组及阿+抑制剂组在造模前1天灌胃阿托伐他汀干预(5 mg/kg,辽宁大连辉瑞制药有限公司,片剂,用温开水2 mL溶解灌胃),1次/天,至造模后3天。假手术组和缺血再灌注组大鼠灌胃2 mL蒸馏水。eIF2a抑制组大鼠在造模前1天灌胃eIF2a酸化特异性抑制剂Salubrinal(11.2 mg/kg,液体针剂,ApexBio Technology LLC公司,Houston,TX,USA)1次/天,至造模后3天[12]。
1.2 TTC染色TTC染色检测大鼠脑缺血组织体积。造模后3天,将大鼠用10%水合氯醛麻醉,用4%多聚甲醛固定并快速断头取脑后,在短暂冰冻后从前至后切成2 mm厚度的脑片,即置于2%TTC 酸盐缓冲液中37 ℃避光恒温解育30 min。PBS固定。拍照,用HMIAS-2000高清晰度彩色图文分析系统(陕西省西安市金马医疗器械有限公司)观察梗死部位,测量梗死体积并计算脑缺血组织体积(占全脑体积的百分比,%)。
1.3 Western blot检测Western blot检测大鼠缺血侧脑组织中PERK、翻译起始因子2a及Caspase-3的表达。造模后3天,将大鼠用10%水合氯醛麻醉并处死,然后将缺血脑组织剪碎各加入一定比例改良RIPA裂解液,在冰浴条件下进行组织匀浆、离心,弃除沉淀,贮存于-80℃中待用。每份样品取样 200 μg,SDS-聚丙烯酰胺凝胶电泳,电压积层胶80 V,分离胶120 V。半干法将蛋白转移至PVDF膜上,加入Western封闭液在室温封闭3 h后,加入—抗兔抗PERK、兔抗eIF2a、兔抗Caspase-3及β-actin多抗(1∶4 000;Santa Cruz Biotechnology,Santa Cruz,CA,USA)4℃孵育过夜,封闭液漂洗,再加HRP标记的山羊抗兔IgG(1∶2 000;Santa Cruz Biotechnology)室温下孵育2 h,摇床杂交1 h,漂洗后放射自显影。测量光密度(OD)值(LS117光密度仪,深圳市金鹏程软件科技有限公司),目的蛋白的表达水平以目的蛋白与β-actin的光密度比值表示。
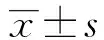
2 结 果
2.1阿托伐他汀增加脑缺血再灌注大鼠脑梗死体积TTC染色结果显示,假手术组大鼠未见脑梗死灶,缺血再灌注组大鼠存在明显脑梗死现象,而与缺血再灌注组相比,阿托伐他汀组及阿+抑制剂组大鼠脑梗死体积明显缩小(P<0.05)(图1)。
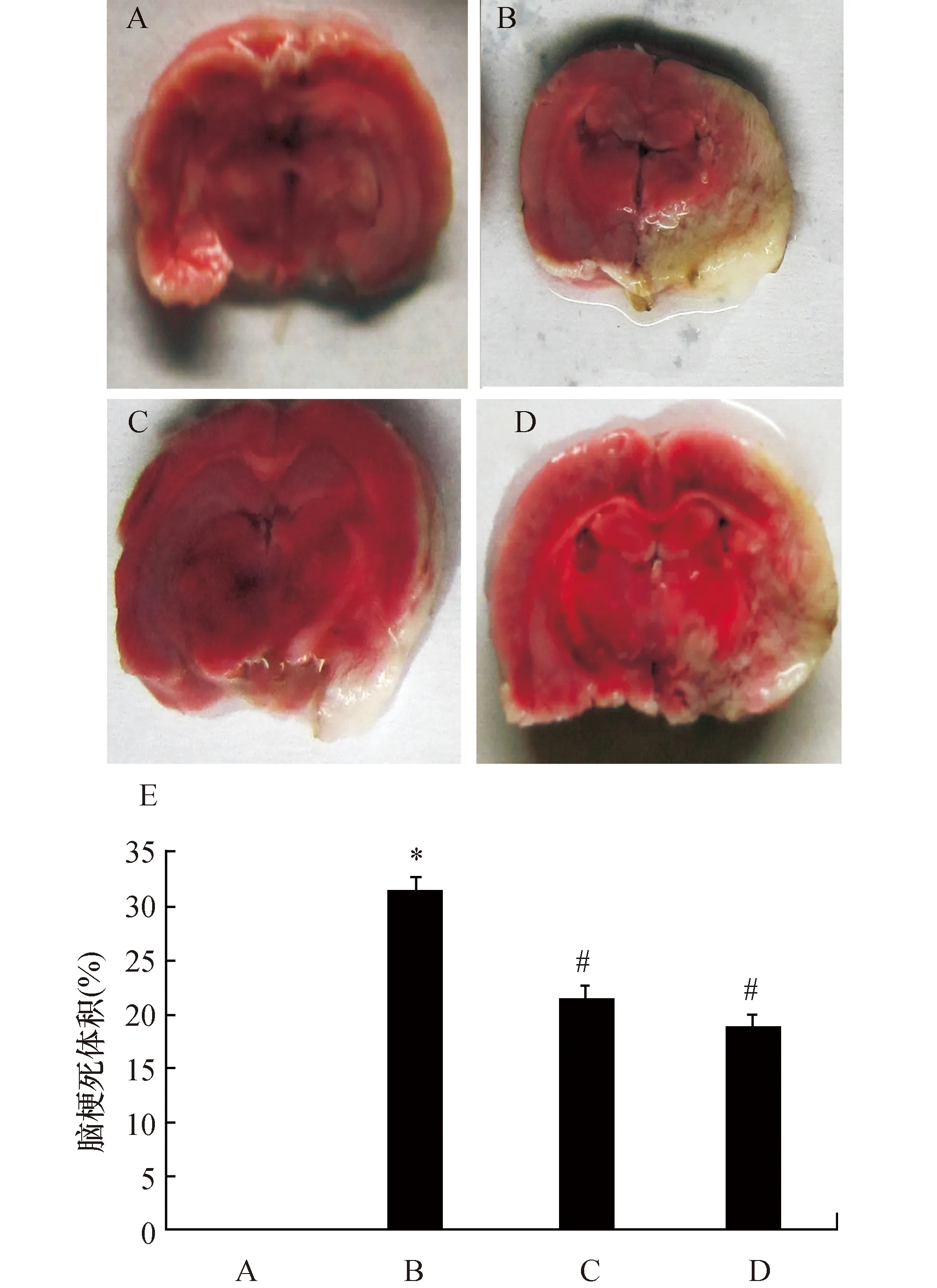
图1 阿托伐他汀对脑缺血再灌注大鼠脑梗死体积的影响大鼠(TTC染色)正常脑组织染成红色,梗死区脑组织染成白色.A:假手术组;B:缺血再灌注组;C:阿托伐他汀+salubrinal抑制剂组;D:阿托伐他汀组;E:大鼠脑梗死体积定量结果. 与假手术组比较,*:P<0.05;与缺血再灌注组比较,#:P<0.05
2.2阿托伐他汀减少脑缺血再灌注大鼠梗死侧脑组织中PERK、Caspase-3和eIF2a表达的变化Western Blot结果显示,与假手术组相比,大鼠缺血再灌注后诱导大量PERK蛋白的表达、Caspase-3活性明显增强及eIF2a蛋白显著磷酸化 (P<0.01)。但阿托伐他汀干预后,缺血再灌注大鼠脑组织中PERK蛋白表达、Caspase-3活性及eIF2a蛋白磷酸化程度明显降低(P<0.05)。而特异性eIF2a酸化抑制剂Salubrinal能显著抑制缺血再灌注大鼠脑组织中eIF2a蛋白磷酸化及Caspase-3活性(P<0.05),但对PERK蛋白表达没有影响(P>0.05,图2)。
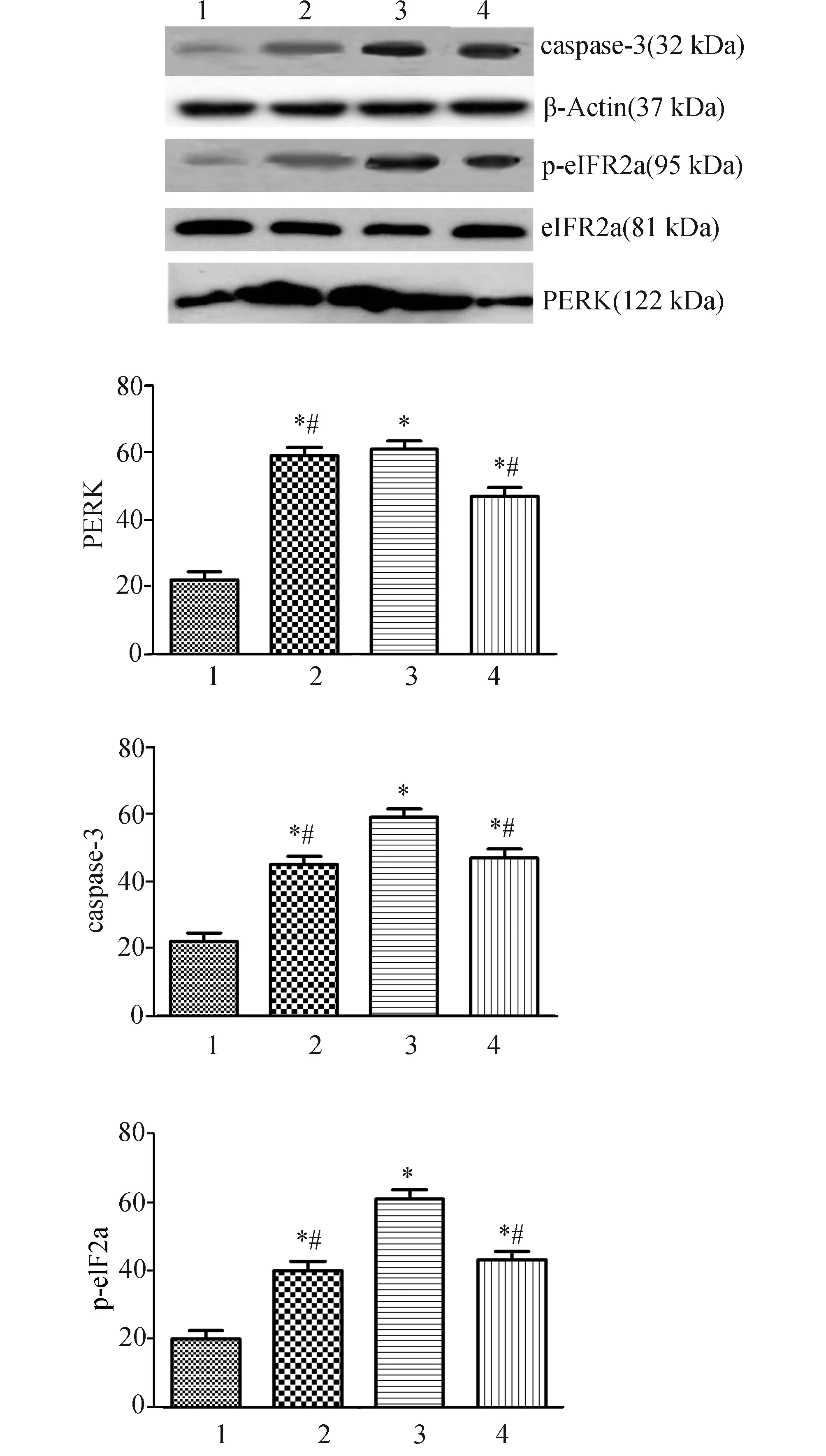
图2 阿托伐他汀对脑缺血再灌注大鼠脑组织中PERK、Caspase-3和eIF2a蛋白表达的影响 蛋白表达水平以其与与β-actin光密度比值表示. 1:假手术组;2:阿托伐他汀+salubrinal抑制组;3:缺血再灌注组;4:阿托伐他汀组. 与假手术组比较,*:P<0.05;与缺血再灌注组比较,#:P<0.05
3 讨 论
蛋白激酶R样内质网激酶是一种丝氨酸苏氨酸激酶,内质网发生应激后,它磷酸化eIF2a[13],从而切断mRNA的翻译,包括ATF4,同时也能诱导UPR相关的基因,降低未折叠蛋白水平。在内质网应激中Caspase-12的过程已有报道[14],导致Caspase-12前体的裂解和激活,进一步激化Caspase-3等因子导致线粒体依赖的细胞死亡,Caspase-3是细胞凋亡关键酶之一,但不能通过自催化或自剪接的方式激活,因而被认为是执行凋亡程序的下游关键因子之一,但具体的PERK/ eIF2a/ Caspase-3通路研究不多。在本研究中TTC染色结果提示缺血再灌注后大鼠存在明显脑梗死现象,阿托伐他汀及Salubrinal干预后大鼠脑梗死体积明显缩小,同时Western Blot测定PERK蛋白的表达增强、eIF2a 明显去磷酸化及Caspase-3活性增加,说明细胞发生凋亡、坏死时激动内质网应激,激活了内质网激酶之一的PERK,也同时使具有mRNA的翻译功能的eIF2a磷酸化加强,切断了蛋白质的翻译过程,并启动了凋亡因子Caspase-3。脑缺血再灌注发生后内质网应激、eIF2a磷酸化及凋亡因子Caspase-3启动三者之间是否有一内在的联系,值得探讨,本实验中设计了eIF2a磷酸化特异性抑制剂Salubrinal组,经Salubrinal抑制后,Caspase-3活性明显下降,从而抑制细胞凋亡,而PERK无影响,推测PERK/eIF2a/Caspase-3通路参与了细胞凋亡途径[15-17]。C/EBP同源蛋白(CHOP)是在内质网触发的凋亡信号中研究最为清楚的分子之一。CHOP抗凋亡主要是ATF4和ATF6依赖的途径调控[18],其另一个重要途径是CHOP介导Bcl-2家族蛋白的转录抑制及激活,从而抑制抗凋亡Bcl-2蛋白的表达[19]。有研究报道PERK/eIF2a/ArF4是CHOP蛋白表达所必需的,PERK信号通路的激活在ERS早期通过抑制蛋白质的合成对细胞起保护作用、促进细胞的生存,随着ERS时间的延长,PERK通过诱导CHOP的表达而促进细胞的凋亡[20-24]。这些前期的研究支持内质网相关的激酶在细胞凋亡中起重要作用,因而内质网应激导致细胞凋亡的途径和因子可能很多,本研究认为内质网应激通过了PERK/ eIF2a/ Caspase-3通路参与了细胞凋亡途径。
氟伐他汀抑制缺氧所导致的巨噬细胞凋亡的机制与N-末端糖调节蛋白(GRP78)有关,内质网应激后PERK通过与GRP78的结合而失去活性[25-27],阿托伐他汀具有调脂、抗炎、抗氧化等作用,显著降低脑血管疾病发生率以及死亡率,阻断HMG-CoA还原成甲基经戊酸(MVA)有关,MVA是焦磷酸拢牛儿酷(GGPP)和焦磷酸法尼醋(FPP)合成的前体,后两者与免疫调节、凋亡有关[28-29]。因而推测阿托伐他汀可能干预内质网应激的凋亡,在本实验中设计了阿托伐他汀组,结果发现阿托伐他汀干预后,PERK蛋白的表达、eIF2a 去磷酸化程度及Caspase-3活性均降低。以往的研究多集中在阿托伐他汀仅通过调脂、抗炎等防止动脉粥样硬化进展等[30-31],在抗凋亡,保护缺血诱导神经细胞死亡研究不多,本研究发现阿托伐他汀可能通过抗凋亡,减轻神经细胞损伤,其机制可能是通过PERK/ eIF2a/ Caspase-3通路参与了细胞凋亡途径。
[1] Gardner BM,Pincus D,Gotthardt K,et al.Endoplasmic reticulum stress sensing in the unfolded protein response[J].Cold Spring Harb Perspect Biol,2013,5(3):a013169.
[2] Zhang R,Piao MJ,Kim KC,et al.Endoplasmic reticulum stress signaling is involved in silver nanoparticles-induced apoptosis[J].Int J Biochem Cell B,2012,44(1):224-232.
[3] Zhang K,Kaufman RJ.The unfolded protein response:a stress signaling pathway critical for health and disease[J].Neurology, 2006,66(2):102-109.
[4] Xu B,Shan M,Wang F,et al.Endoplasmic reticulum stress signaling involvement in manganese-induced nerve cell damage in organotypic brain slice cultures[J].Toxicol Lett,2013,222(3):239-246.
[5] Oyadomari S,Mori M.Roles of CHOP/GADD153 in endoplasmic reticulum stress[J].Cell Death Differ, 2004,11(4):381-389.
[6] Senkal CE,Ponnusamy S,Bielawski J,et al.Antiapoptotic roles of ceramide-synthase-6-generated C16-ceramide via selective regulation of the ATF6/CHOP arm of ER-stress-response pathways[J].FASEB J, 2010,24(1):296-308.
[7] Hetz C,Martinon F,Rodriguez D,et al.The unfolded protein response:integrating stress signals through the stress sensor IRE1α[J].Physiol Rev, 2011,91(4):1219-1243.
[8] Mandl J,Mészáros T,Bánhegyi G,et al.Minireview:endoplasmic reticulum stress:control in protein,lipid,and signal homeostasis[J].Mol Endocrinol,2013,27(3):384-393.
[9] Sone M,Zeng X,Larese J,et al.A modified UPR stress sensing system reveals a novel tissue distribution of IRE1/XBP1 activity during normal Drosophila development[J].Cell Stress Chaperones,2013, 18(3):307-319.
[10] 唐丹,陈晓平,陈国柱,等.氟伐他汀对动脉粥样硬化斑块中巨噬细胞凋亡的影响[J].西部医学,2008,20(1):10-11.
[11] 崔淑华,庹勤慧.动脉粥样硬化中巨噬细胞凋亡与内质网应激机制[J].国际病理科学临床杂志,2011,31(4):315-318.
[12] Teng Y,Gao M,Wang J,et al.Inhibition of eIF2α dephosphorylation enhances TRAIL-induced apoptosis in hepatoma cells[J].Cell Death Dis,2014, 5:e1060.
[13] Lee SK,Kim YS.Phosphorylation of eIF2α attenuates statin-induced apoptosis by inhibiting the stabilization and translocation of p53 to the mitochondria[J].Int J Oncol,2013,42(3):810-816.
[14] Ma Y,Hendershot LM.The unfolding tale of the unfolded protein response[J].Cell,2001,107(7):827-830.
[15] Morishima N,Nakanishi K,Takenouchi H,et al.An endoplasmic reticulum stress-specific Caspase cascade in apoptosis.Cytochrome c-independent activation of Caspase-9 by Caspase-12[J].J Biol Chem,2002,277(4):34287-34294.
[16] Li Z,Jo J,Jia JM,et al.Caspase-3 activation via mitochondria is required for long-term depression and AMPA receptor internalization[J].Cell,2010,141(5):859-871.
[17] Walter P,Ron D.The unfolded protein response:from stress pathway to homeostatic regulation[J].Science,2011,334(6059):1081-1086.
[18] Zhu H,Zhu H,Xiao S,et al.Activation and crosstalk between the endoplasmic reticulum road and JNK pathway in ischemia-reperfusion brain injury[J].Acta Neurochir (Wien),2012, 154(7):1197-1203
[19] Weston RT,Puthalakath H.Endoplasmic reticulum stress and BCL-2 family members[J].Adv Exp Med Biol,2010,687:65-77.
[20] Porter AG,Jänicke RU.Emerging roles of Caspase-3 in apoptosis[J].Cell Death Differ,2010, 6(2):99-104.
[21] Salakou S,Kardamakis D,Tsamandas,et al.Increased Bax/Bcl-2 ratio up-regulates Caspase-3 and increases apoptosis in the thymus of patients with myasthenia gravis[J].In Vivo,2007,21(1):123-132.
[22] Trivedi R,Maurya R,Mishra DP.Medicarpin,a legume phytoalexin sensitizes myeloid leukemia cells to TRAIL-induced apoptosis through the induction of DR5 and activation of the ROS-JNK-CHOP pathway[J].Cell Death Dis,2014,165:e1465.
[23] Park HJ,Park SJ,Koo DB,et al.Progesterone production is affected by unfolded protein response (UPR) signaling during the luteal phase in mice[J].Life Sci,2014,113(1-2):60-67.
[24] Wang M,Meng X,Yu Y,et al.Elatoside C protects against hypoxia/reoxygenation-induced apoptosis in H9c2 cardiomyocytes through the reduction of endoplasmic reticulum stress partially depending on STAT3 activation[J].Apoptosis,2014,19(12):1727-1735.
[25] Jia F,Wu C,Chen Z,et al.Atorvastatin inhibits homocysteine-induced endoplasmic reticulum stress through activation of AMP-activated protein kinase[J].Cardiovasc Ther,2012,30(6):317-325.
[26] 杨帆,马允,昂文平,等.针刺对戊四唑诱发惊厥大鼠海马葡萄糖调节蛋白78和C/EBP同源蛋白表达的影响[J].针刺研究,2014,39(4):267-271.
[27] Yang X,Xu H,Hao Y,et al.Endoplasmic reticulum oxidoreductin 1α mediates hepatic endoplasmic reticulum stress in homocysteine-induced atherosclerosis[J].Acta Biochim Biophys Sin (Shanghai),2014,46(10):902-910.
[28] Chen JC,Wu ML,Huang KC,et al.HMG-CoA reductase inhibitors activate the unfolded protein response and induce cytoprotective GRP78 expression[J].Cardiovasc Res,2008,80(1):138-150.
[29] Qian J,Keyes KT,Long B,et al.Impact of HMG-CoA reductase inhibition on oxidant-induced injury in human retinal pigment epithelium cells[J].J Cell Biochem,2011,112(9):2480-2489.
[30] Gleissner CA,Leitinger N,Ley K.Effects of native and modified low-density lipoproteins on monocyte recruitment in atherosclerosis[J].Hypertension,2007,50(2):276-283.
[31] Vilahur G,Casaní L,Pea E,et al.Induction of RISK by HMG-CoA reductase inhibition affords cardioprotection after myocardial infarction[J].Atherosclerosis,2009,206(1):95-101.
TheStudyofPERK/eIF2aPathwayandtheExpressionofCaspase-3inIschemia-reperfusionRatsandAtorvastatinIntervention
PENG Wenjuan,YANG Jianwen,LIU Xiangyu,et al
(MawangduiHospitalAffilatedtoUniversityofSouthChina,Changsha,Hunan410015,China)
ObjectiveTo study the PERK/eIF2a pathways and Caspase 3 in the mechanism of action of ischemia reperfusion injury in rats and the effect of atorvastatin.MethodsProduce the ischemia reperfusion model rats by Middle cerebral artery embolism method which were divided into the ischemia reperfusion group,control group,atorvastatin intervention group and eIF2a suppression group.To observe the changes of ischemic brain,specimens were treated with TTC staining,the PERK,Caspase-3 protein expression and protein phosphorylation eIF2a were detected by western-blot.Results
Compared with the control group,after ischemia reperfusion,PERK protein expression and protein phosphorylation eIF2a increased,the expression of Caspase 3 was enhanced,and the expression of PERK protein and phosphorylation eIF2a was relieved after atorvastatin intervention.The expression of Caspase-3 and phosphorylation eIF2a was inhibited by inhibitor Salubrinal,which had no effect no PERK.Large cerebral infarction in ischemia reperfusion group was visible by TTC staining after ischemia reperfusion.Cerebral infarction volume was significantly narrowed by atorvastatin intervention or inhibitor Salubrinal.ConclusionCell apoptosis was related to the PERK/ eIF2a /Caspase 3 pathways after endoplasmic reticulum stress,atorvastatin can reduce the damage of cerebral ischemia reperfusion.
protein kinase-like ER kinase; EIF2a; endoplasmic reticulum stress; Caspase-3; ischemia reperfusion; atorvastatin
R743.3
A
10.15972/j.cnki.43-1509/r.2016.01.005
2015-10-08;
2015-11-26
湖南省卫生计生委资助项目(B2015-73).
*通讯作者,E-mail:csyang904@126.com.
朱雯霞)
猜你喜欢
天津医科大学学报(2021年3期)2021-07-21
世界科学技术-中医药现代化(2021年12期)2021-04-19
现代临床医学(2021年1期)2021-01-26
山东医药(2021年28期)2021-01-11
昆明医科大学学报(2020年11期)2020-12-28
中成药(2018年4期)2018-04-26
中国组织化学与细胞化学杂志(2017年1期)2017-06-15
中国医药生物技术(2015年4期)2015-12-26
中国康复理论与实践(2015年10期)2015-12-24
中国体外循环杂志(2015年3期)2015-12-08
