
吡唑并[3,4-b]吡啶衍生物对A1腺苷受体拮抗活性的定量构效关系
2016-12-20 02:22刘慧群
宿州学院学报 2016年11期
陈 新,刘慧群
皖西学院材料与化工学院,安徽六安,237012
吡唑并[3,4-b]吡啶衍生物对A1腺苷受体拮抗活性的定量构效关系
陈 新,刘慧群
皖西学院材料与化工学院,安徽六安,237012

定量构效关系(QSAR);启发式方法;吡唑并[3,4-b]吡啶衍生物;pKi
寻找高效选择性的A1腺苷受体拮抗剂已成为近年来的研究热点之一[1-5]。为此,人们合成了许多含氮杂环化合物吡唑并[3,4-b]吡啶衍生物、吡唑并[3,4-b]嘧啶衍生物和吡唑并[3,4-b]吡啶酮衍生物。Paola Fossa等人[1-2]用偏最小二乘法(PLS)研究了吡唑并[3,4-b]吡啶衍生物与A1腺苷受体结合常数的构效关系,得到了15参数模型,但是该模型对吡唑并[3,4-b]吡啶衍生物类似物结合常数的预测结果与实验值偏差很大。鉴于CODESSA软件[6]在预测化合物理化性质和生物学性质方面取得很好的效果[7-14],本文采用该软件进行理论计算,以期得到吡唑并[3,4-b]吡啶衍生物与A1腺苷受体结合常数的理想的预测模型。
1 计算方法

2 结果与讨论
计算得到的六参数和七参数模型分别表示为EQ-6和EQ-7,方程中的变量含义如表2所示:
pKi=(-21.02±1.767)BO-Cmax-(4690.1
±537.1)PC-Nmax+(0.0217±0.0028)ESP-HDSA+(7.6906±1.5239)FNSA-2+(1.1754±0.2284)ESPmin-(35.060±9.1307)ESP-FHBCA-(289.59±36.904)
EQ-6
pKi=(-21.672±3.3076)BO-Cmax-(3789.7±466.30)PC-Nmax+(0.027597±0.0033222)ESP-HDSA+(7.0889±1.4223)FNSA-2-(57.812±6.9588)NRI-Nmin-(58.301±11.661)ESP-FHBCA+(1780.9±493.14)HACA-2-(221.74±28.374)
EQ-7
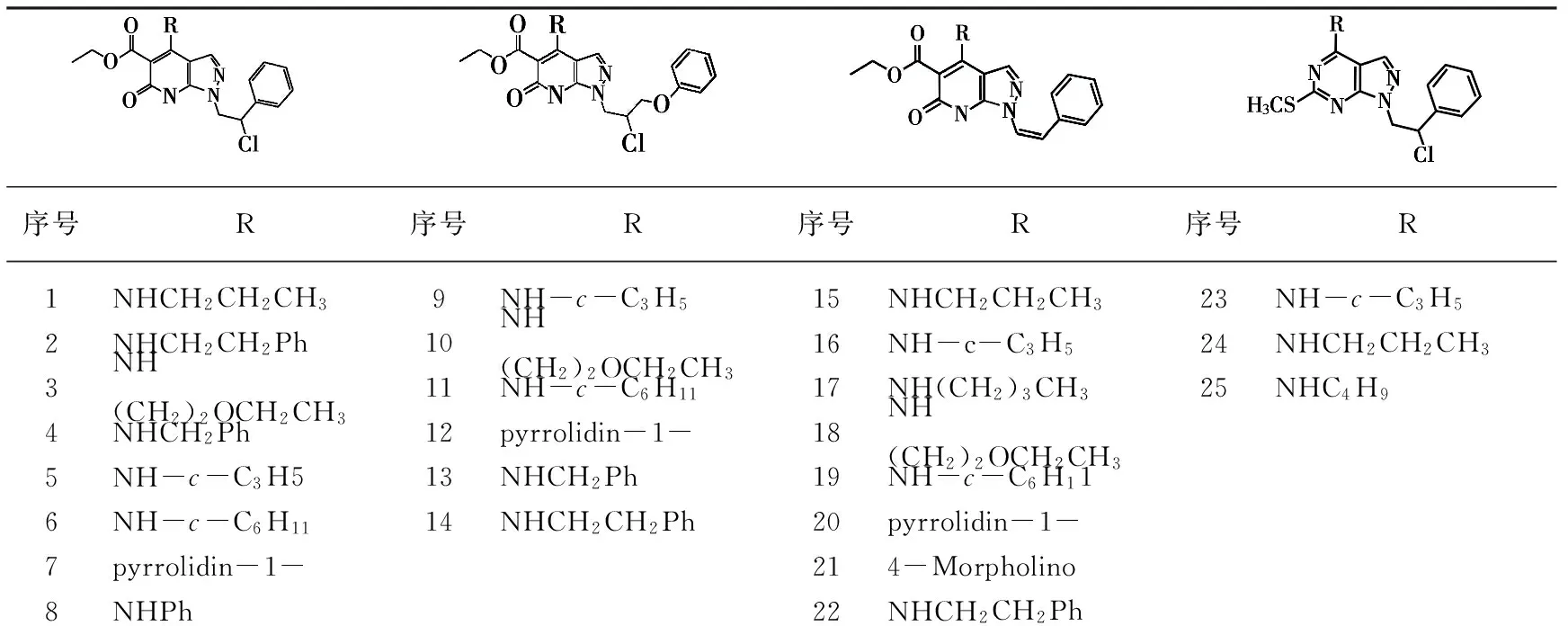
序号R序号R序号R序号R1NHCH2CH2CH39NH-c-C3H515NHCH2CH2CH323NH-c-C3H52NHCH2CH2Ph10NH(CH2)2OCH2CH316NH-c-C3H524NHCH2CH2CH33NH(CH2)2OCH2CH311NH-c-C6H1117NH(CH2)3CH325NHC4H94NHCH2Ph12pyrrolidin-1-18NH(CH2)2OCH2CH35NH-c-C3H513NHCH2Ph19NH-c-C6H116NH-c-C6H1114NHCH2CH2Ph20pyrrolidin-1-7pyrrolidin-1-214-Morpholino8NHPh22NHCH2CH2Ph

序号R1R序号R1R26CH3NH-c-C3H548CH(CH3)2NH-c-C3H527CH3NHCH2CH2CH349CH(CH3)2NHCH2CH2CH328CH3pyrrolidin-1-50CH(CH3)2pyrrolidin-1-29CH34-Morpholino51CH(CH3)24-Morpholino30CH3NHCH2Ph52CH(CH3)2NHCH2Ph31CH3NHCH2CH2Ph53CH(CH3)2NHCH2CH2Ph32CH3NHCH2CH2-p-C6H4-CH354CH(CH3)2NHCH2CH2-p-C6H4-CH333CH3NHCH2CH2-p-C6H4-OCH355CH(CH3)2NHCH2CH2-p-C6H4-OCH334C2H5NHCH2CH2-p-C6H4-CH356CH(CH3)2NHCH2CH2-o-C6H4-Cl35C2H5NHCH2CH2-p-C6H4-OCH357CH(CH3)2NHCH2CH2-m-C6H4-Cl36C2H5NHCH2CH2-o-C6H4-F58CH(CH3)2NHCH2CH2-p-C6H4-Cl37C2H5NHCH2CH2-m-C6H4-F59C4H9NH-c-C3H538C2H5NHCH2CH2-p-C6H4-F60C4H9NHCH2CH2CH339C2H5NHCH2CH2-o-C6H4-Cl61C4H9pyrrolidin-1-40C2H5NHCH2CH2-m-C6H4-Cl62C4H94-Morpholino41C2H5NHCH2CH2-p-C6H4-Cl63C4H9NHCH2Ph42C3H7NH-c-C3H564CH(CH3)C2H5NH-c-C3H543C3H7NHCH2CH2CH365CH(CH3)C2H5NHCH2CH2CH344C3H7pyrrolidin-1-66CH(CH3)C2H5NHCH2CH2Ph45C3H74-Morpholino67NHCH2CH2Ph46C3H7NHCH2Ph68NHCH2CH2-p-C6H4-CH347C3H7NHCH2CH2Ph
图1 68种模型化合物的结构图

表1 六参数和七参数模型的结合强度预测值与实验值
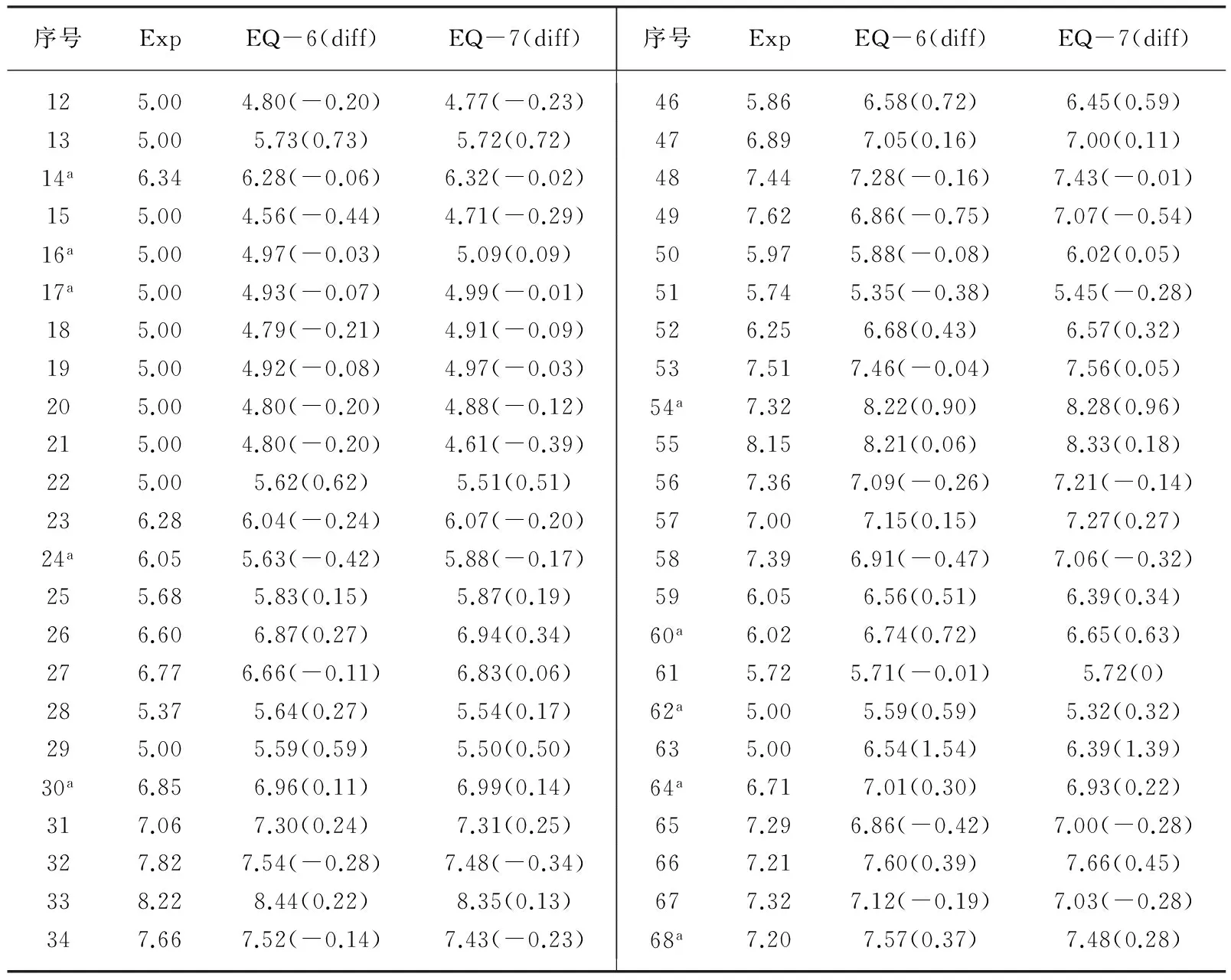
续表
注:序号中标注a的化合物为测试集化合物,Exp为实验值,EQ-6(diff)表示EQ-6的预测值及其误差。

表2 QSAR模型中描述符的含义
从方程EQ-6和EQ-7可以看出,两者都共同使用了BO-Cmax(碳原子的最大键级)、PC-Nmax(氮原子的最大部分电荷)、ESP-HDSA(氢键供体氢原子表面静电势)、FNSA-2(电荷加权部分负电荷表面积的分数)和ESP-FHBCA(面积加权氢键供体原子静电势)。不同之处在于EQ-6使用了参数ESPmin(分子表面的最小静电势),而EQ-7使用了参数NRI-Nmin(氮原子的最小亲核反应指数)和HACA-2(氢键受体原子的面积加权表面电荷)。从中可以看出,影响吡唑并[3,4-b]吡啶衍生物及其类似物与A1腺苷受体结合强度的主要因素可以分为碳原子的最大键级、氢键相互作用和静电相互作用三种类型。吡唑并[3,4-b]吡啶衍生物及其类似物中氮原子作为氢键受体,其带电状况既影响氢键作用的强度,也影响静电作用的强度。
从预测结果来看,EQ-6和EQ-7对惰性化合物(pKi指定为5.00)预测误差最大的为化合物63,绝对误差分别为1.54和1.39,相对误差分别为30.8%和27.8%;对于活性化合物预测误差最大的为化合物4,绝对误差分别为0.99和0.89,相对误差分别为15.2%和13.7%。除此之外,EQ-6和EQ-7对训练集和测试集化合物(不论是惰性化合物还是活性化合物)预测值的相对误差均在0(化合物43)~11.9%(化合物41)范围之内,而且预测结果非常接近,进一步增加预测模型的参数意义不大。因此,EQ-6可以较好地吡唑并[3,4-b]吡啶衍生物及其类似物与A1腺苷受体结合强度。
3 结 论

[1]Bondavalli F,Botta M,Bruno O,et al.Synthesis,molecular modeling studies,and pharmacological activity of selective A1 receptor antagonists[J].Journal of Medicinal Chemistry,2002,45(22):4875-4887
[2]Manetti F,Schenone S,Bondavalli F,et al.Synthesis and 3D QSAR of new pyrazolo[3,4-b]pyridines:potent and selective inhibitors of A1 adenosine receptors.[J].Journal of Medicinal Chemistry,2005,48(23):7172-85
[3]Fan L,Yao C,Shu M.Three-component synthesis of new o-hydroxyphenyl-substituted pyrazolo[3,4-b]pyridines promoted by FeCl3[J].Heterocyclic Communications,2016,22(2):63-67
[4]Squarcialupi L,Falsini M,Catarzi D,et al.Exploring the 2-and5-positions of the pyrazolo[4,3-d]pyrimidin-7-amino scaffold to target human A 1,and A2A,adenosine receptors[J].Bioorganic & Medicinal Chemistry,2016,24(12):2794-2808
[5]Elgendy H.Pyrazolopyrimidines:Synthesis,Chemical reactions and Biological activity[J].International Journal of Advanced Research,2014,2(7):474-564
[6]University of Florida.Codessa Pro software[CP/OL].[2016-7-8].http://www.codessa-pro.com
[7]Girgis A S,Panda S S,Aziz M N,et al.Rational design,synthesis,and 2D-QSAR study of anti-oncological alkaloids against hepatoma and cervical carcinoma[J].Rsc Advances,2015,36:28554-28569
[8]Devillers J,Doucet-Panaye A,Doucet J P.Structure-activity relationship(SAR) modelling of mosquito larvicides.[J].Sar & Qsar in Environmental Research,2015,26(4):1-16
[9]Irfan M,Aneja B,Yadava U,et al.Synthesis,QSAR and anticandidal evaluation of 1,2,3-triazoles derived from naturally bioactive scaffolds[J].European Journal of Medicinal Chemistry,2015,93:246-254
[10]Pourbasheer E,Aalizadeh R,Ardabili J S,et al.QSPR study on solubility of some fullerenes derivatives using the genetic algorithms-Multiple linear regression[J].Journal of Molecul- ar Liquids,2015,204:162-169
[11]Srour A M,El-Karim S S A,Saleh D O,et al.Rational design,synthesis and 2D-QSAR study of novel vasorelaxant active benzofuran-pyridine hybrids[J].Bioorganic & Medicinal Chemistry Letters,2016,26(10):2557-2561
[12]Hui W,Nguyen T T H,Li S,et al.Quantitative structure-activity relationship of antifungal activity of rosin derivatives[J].Bioorganic & Medicinal Chemistry Letters,2015,25(2):347-354
[13]Girgis A S,Saleh D O,George R F,et al.Synthesis,bioassay,and QSAR study of bronchodilatory active 4 H -pyrano[3,2- c ]pyridine-3-carbonitriles[J].European Journal of Medicinal Chemistry,2015,89:835-843
[14]Braga R C,Alves V M,Silva M F,et al.Tuning HERG out:antitarget QSAR models for drug development[J].Current Topics in Medicinal Chemistry,2014,14(11):1399-415
[15]University of Florida.AMPAC software[CP/OL].[2016-7-8].http://www.semichem.com/ampac/
(责任编辑:汪材印)
2016-05-16
安徽省自然科学基金“载银多孔配位聚合物的设计合成及其抗菌与缓释性能研究”(1308085ME57);皖西学院大学生研究性学习项目“多孔纳米Fe3O4@SiO2固定组氨酸配合物催化氧化环己烷的性能研究”(wxxyx2016013)。
陈新(1972-),安徽六安人,博士,副教授,主要研究方向:有机化学和药物化学。
10.3969/j.issn.1673-2006.2016.10.031
O06
A
1673-2006(2016)11-0118-04
猜你喜欢
检察风云(2022年5期)2022-04-05
云南化工(2021年10期)2021-12-21
今日农业(2021年2期)2021-11-27
今日农业(2020年23期)2020-12-31
化工学报(2020年4期)2020-05-28
今日农业(2019年11期)2019-08-13
国外医药(抗生素分册)(2016年3期)2016-07-12
合成化学(2015年1期)2016-01-17
山东医药(2015年16期)2016-01-12
无机化学学报(2014年7期)2014-02-28
