
青岛大气气溶胶中微量元素溶解度及影响因素
2016-12-20 08:16石金辉1贲孝宇高会旺1姚小红1中国海洋大学海洋环境与生态教育部重点实验室山东青岛66100中国海洋大学环境科学与工程学院山东青岛66100中国海洋大学海洋环境学院山东青岛66100
中国环境科学 2016年11期
朱 敏,石金辉1,*,贲孝宇,仇 帅,高会旺1,,姚小红1,(1.中国海洋大学,海洋环境与生态教育部重点实验室,山东 青岛 66100;.中国海洋大学环境科学与工程学院,山东 青岛 66100;.中国海洋大学海洋环境学院,山东 青岛 66100)
青岛大气气溶胶中微量元素溶解度及影响因素
朱 敏2,石金辉1,2*,贲孝宇3,仇 帅2,高会旺1,2,姚小红1,2(1.中国海洋大学,海洋环境与生态教育部重点实验室,山东 青岛 266100;2.中国海洋大学环境科学与工程学院,山东 青岛 266100;3.中国海洋大学海洋环境学院,山东 青岛 266100)
利用2012年12月在青岛采集的31个总悬浮颗粒物(TSP)样品,分析其中11种微量元素的总浓度和溶解态浓度,讨论了沙尘负载、气溶胶来源、酸过程及天气条件对微量元素溶解度的影响.结果表明,Fe和Al的溶解度<5%,Pb、Ba、Bi的为10%左右,Cu、V、Cd、Mn的为20%~30%,Zn和As的约为40%.随着沙尘负载量的增加,气溶胶中微量元素溶解度呈规律性递减.气团后向轨迹聚类分析和正矩阵因子分析(PMF)结果显示,受人为源影响的气溶胶中微量元素溶解度明显高于受沙尘源影响的,受二次生成源和海洋源影响的的气溶胶中微量元素溶解度明显高于受土壤源影响的.大气酸过程是造成这些差异的主要原因.相关性分析表明,S O42-、NO3-和有机酸等酸组分均对微量元素溶解度有一定影响.霾天时气溶胶中微量元素溶解度明显低于雾天的,其原因为酸组分在较高的相对湿度下更能促进微量元素溶解度的提高.
微量元素;气溶胶;溶解度;酸化过程;雾霾
大气气溶胶中的微量元素通过干湿沉降进入海洋,对维持海水中Fe、Mn、Zn等生物必需的微量元素浓度起着重要作用,对海洋生物的生长有着重要影响[1-5].沉降入海的微量元素的生物可利用性很大程度上取决于其溶解态的浓度而非总浓度,溶解态浓度占总浓度的百分比为微
量元素的溶解度.关于微量元素溶解度的研究在世界各地已有一些报道,如欧洲[6-7]、中国上海及东海[8-9]、大西洋[10]及太平洋海域[11]等.但不同研究所报道的微量元素溶解度变化范围很大,这可能是由于影响溶解度因素复杂的缘故[12].已有研究显示,大气输入海洋的微量元素溶解度受到大气过程和海洋过程的双重控制[13].仅大气过程即包括气溶胶的来源和化学组成[7,14]、气溶胶的粒径谱分布[15]、大气酸化过程[16-17]、云过程和老化过程[18]等.
青岛位于山东半岛南端,东南濒临东亚季风发达的黄海,处于亚洲沙尘源区和华北城市群的下风带,是亚洲大陆气溶胶向西北太平洋传输的重要通道.在青岛开展大气气溶胶中微量元素溶解度的研究,不仅有助于了解人为活动对亚洲大陆气溶胶中微量元素溶解度的影响,而且有助于评价其沉降对中国近海初级生产力的影响.因此,本文利用2012年12月在青岛连续采集的31个总悬浮颗粒物(TSP)样品,分析其中 11种微量元素总的和溶解态的浓度,以认识黄海近岸大气气溶胶中微量元素的溶解度及其主控因子.
1 样品采集与分析
1.1 样品采集
采样地点设在青岛市市南区中国海洋大学鱼山校区达尔文楼顶(36°06′N,120°33′E,海拔约65m),距离海岸线<1km,附近无明显工业污染源.采样时间为2012年12月,每天连续采集,共采集了31个TSP样品,每个样品于当日12:00开始采集至次日12:00结束.采样器为KC-1000型大流量总悬浮颗粒物采样器(青岛崂山电子公司),采样流量为1.05m3/min.采样膜为Whatman 41纤维滤膜(预先经10%HCl、1%HNO3及Milli-Q超纯水清洗).采样结束后,在洁净室中小心取下滤膜,将滤膜对折放入洁净的聚乙烯封口袋中于-20℃下冷冻保存至分析.
采用激光粒子计数器(HHPC-6, ARTI)观测采样同期大气颗粒物数浓度谱,该仪器共6个粒径通道,分别为0.3、0.5、0.7、1.0、2.0和5.0µm,采样流速为2.83L/min,每15min测定1次数浓度.
1.2 样品预处理
取一定面积样品膜用 2mL浓 HNO3和0.5mL浓HF于聚四氟乙烯高效消解罐中180℃下消解至完全,取出消解罐内胆于 180℃加热赶酸后加2% HNO3溶解残渣并定容,用于样品中微量元素总浓度的分析;取一定面积样品膜用20mL Milli-Q水(≥18.2MΩ·cm)在 0℃下超声波萃取1h,萃取液经0.45µm滤膜过滤后酸化、定容,用于溶解态微量元素分析;取一定面积样品膜用15mL Milli-Q水0℃下超声波萃取30min,萃取液经 0.45µm滤膜过滤后定容,用于样品中无机、有机离子分析.上述处理后的样品溶液转移至样品瓶中4℃下保存,并在2d内完成分析.
1.3 样品分析
样品中Al、Fe、Pb、Ba、Bi、Cu、V、Cd、Mn、Zn和As等11种微量元素采用电感耦合等离子体质谱仪(Agilent 7500c ICP-MS)进行分析
[19], Na+、NH4+、K+、Mg2+、Ca2+等阳离子和Cl-、NO3
-、SO42-、乙二酸(C2O42-)等阴离子采用离子色谱仪(Dionex ICS-3000)进行分析[20].每分析10个样品加测一与样品浓度接近的标准溶液,以确保分析结果准确.
1.4 气溶胶粒子比表面积计算
气溶胶粒子的比表面积(S)用以下公式计算:

式中:Si、Ai、Mi分别为第i个气溶胶样品采集期间颗粒物的比表面积(µm2/µg)、表面积(µm2/m3)和质量浓度(µg/m3);Ci,j为相应样品采集期间第j级颗粒物平均数浓度,个/m3;dj为第j级颗粒物空气动力学直径,µm.
2 结果与讨论
2.1 青岛气溶胶中微量元素的总浓度及溶解度
青岛大气气溶胶中微量元素总浓度以Al和Fe浓度最高,平均分别为3560,3248ng/m3,其次是Zn、Pb、Mn和Ba,分别为230,99,63,58ng/m3, Cu、As和V的浓度较低,均低于50ng/m3, Cd和Bi
的浓度最低,低于 5ng/m3.溶解态的微量元素以Zn的平均浓度最高,为105ng/m3,其次是Al、Fe和Mn,平均分别为59,43,19ng/m3,其他元素的溶解态浓度均低于 10ng/m3,Cd和 Bi的低于1ng/m3(表1).

表1 青岛大气气溶胶中微量元素总浓度、溶解态浓度及溶解度Table 1 Total and soluble concentrations of trace elements and solubilities in aerosols collected in Qingdao
尽管Al和Fe是气溶胶中总浓度最高的微量元素,但其溶解度却是最低的.Fe溶解度均在5%以下,Al的溶解度除在12月12、14和16日为10%~18%外,其他样品基本在5%以下(图1).Pb、Ba和Bi的溶解度除在12月12日为20%左右外,多数样品Pb溶解度为5%,Ba和Bi的为10%左右;Cu的溶解度基本在10%~30%之间,平均约为20%;V、Cd和 Mn的溶解度较高,基本在10%~50%之间,平均约为30%;Zn和As的溶解度最高,多数样品中的为20%~60%,均值约为40%.与 Hsu等[9]报道的冬季东海气溶胶中微量元素溶解度相比,尽管青岛大气气溶胶中微量元素总浓度除 Cu外均高于东海气溶胶中的,但溶解度均低于东海的,其中Al、Fe、Ba、Mn和 V等主要来自地壳源贡献的微量元素溶解度略低于东海的,但Cu、Zn、Pb、As和Cd等主要受人为源影响的元素溶解度则明显低于东海的.这可能是由于到达海上的自然源气溶胶经过长距离的输运更有机会与酸性污染物混合从而使其中地壳源元素的溶解度有所提高,而东海气溶胶中人为源元素溶解度明显高于青岛的可能是由于东海濒临我国经济发达的上海等华东地区受到人为污染的影响更显著[21].
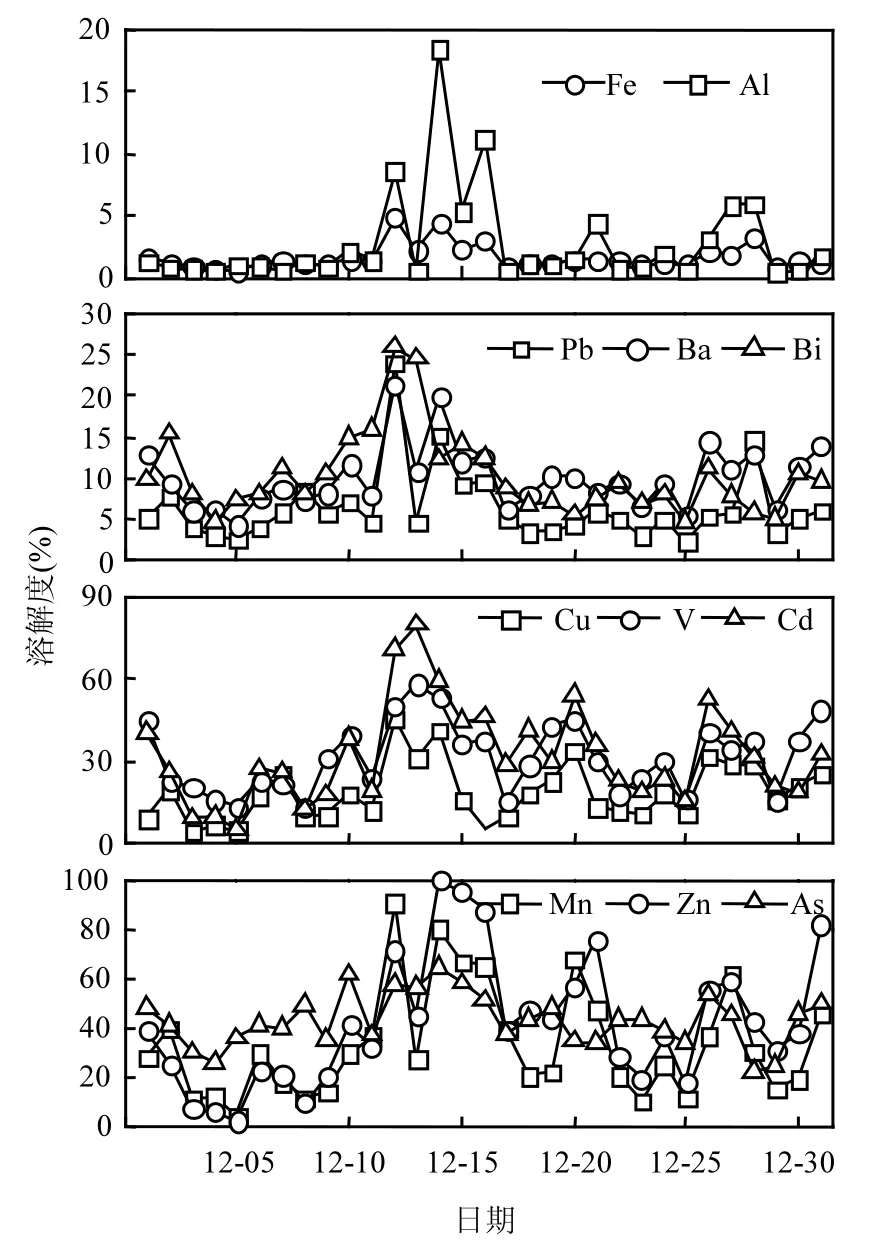
图1 青岛大气气溶胶中微量元素的溶解度Fig.1 Temporal variations of solubility of trace elements in aerosols collected in Qingdao
对比分析微量元素的总浓度和溶解度,发现微量元素溶解度高时,总浓度往往较低,而溶解度低时,总浓度往往较高,如Al、Fe和Mn的最高溶解度分别为18.5%、4.9%和90.6%,相对应的总浓度分别为962、1214和25.7ng/m3,接近最小浓度;3者的最低溶解度分别为 0.46%,0.57%,4.8%,对应的总浓度分别为 5364,5114,89.1ng/m3,接近最大浓度.统计分析显示,除Pb和Bi外,微量元素溶解度均与总浓度呈现显著负相关关系(r=-0.4~-0.7,P<0.05).Baker等[22]在研究大西洋受沙哈拉沙尘影响的气溶胶时也发现Fe、Al、Mn等微量元素的溶解度随总浓度的增加呈规律性减小,并认为这种关系与气溶胶的沙尘负载量或颗粒物
的大小有关.
2.2 沙尘负载对微量元素溶解度的影响
已有研究表明微量元素溶解度与沙尘负载量存在一定关系[13].假设青岛大气气溶胶中的Al全部来自沙尘源,其在地壳中的丰度约为8%[23],由Al总浓度可以反推气溶胶中沙尘负载量(md).结果显示,青岛气溶胶中微量元素溶解度随沙尘负载量的增大均呈规律性递减,拟合曲线的相关系数为 0.35~0.78(图 2,举例给出了气溶胶中含量相对较高的 6种元素的溶解度和沙尘负载量的关系).溶解度与沙尘负载量之间的关系与气溶胶粒子相对表面积的大小有关[15].气溶胶在长距离传输过程中,由于重力原因粒径较大的粒子会优先沉降下来,因而随着传输时间和距离的增加气溶胶粒子的比表面积逐渐增大,这为气溶胶中微量元素与大气中酸性污染物反应提供了更大面积的载体,从而更有利于其溶解度提高.利用观测期间颗粒物数浓度谱数据和颗粒物质量浓度,计算每个样品采集期间气溶胶粒子的比表面积,发现青岛气溶胶中微量元素的溶解度均与气溶胶粒子的比表面积有一定的相关关系(r>0.4, P<0.05),表明微量元素溶解度与颗粒物的大小有关.
气溶胶中微量元素的溶解度受物理和化学因素的共同影响[13].Shi等[24]通过对比不同粒径颗粒物中Fe和Al溶解度的模拟值和实测值,发现模拟值总是比实测值小1~2个数量级,这说明只考虑颗粒物大小这一物理因素的影响只能对溶解度的变化做出一小部分解释,而未经历大气化学过程的沙尘颗粒中Fe和Al的溶解度很难超过1%[12,24].
2.3 气溶胶来源对微量元素溶解度的影响
2.3.1 气团后向轨迹分析 气溶胶样品采集期间气团的后向轨迹可以反应其来源.根据采样期间气团后向轨迹的聚类分析将采集的31个样品分为受华北城市人为源(AS)和北方沙尘源(DS)影响的 2组[19].AS组中微量元素溶解度均大于DS组的,且增幅基本在30%以上(表2).在英国伯明翰[7]、爱丁堡[14]以及中国东海[9]气溶胶微量元素溶解度的研究中,也发现来自非沙尘源样品中微量元素溶解度高于沙尘源样品的.造成这种差异的原因可能是由于不同来源气溶胶中微量元素的初始来源或组成不同,也可能是经历了不同的大气化学过程.

图2 青岛气溶胶中微量元素溶解度与沙尘负载量关系Fig.2 Correlations of trace element solubility versus dust loading in aerosols collected from Qingdao
富集因子(EF)常用来判断气溶胶中元素的来源,以Al为地壳源参比元素,计算AS组和DS组气溶胶中微量元素的富集因子.可以看出(图3),无论是主要来自地壳源的Al、Fe、Mn、V、Ba(EF<4),还是受地壳源和人为源共同影响的Cu(EF≈10),以及主要来自人为源的Zn、As、Pb、Cd、Bi(EF≥100),其EF值在AS和DS组气溶胶中均基本相当,这说明 2组样品中元素的初始来源或组成相似.因此,2组样品中微量元素溶解度的差异并非是元素的初始来源或组成不同造成的,而可能是气溶胶在大气传输过程中经历了不同的大气过程.研究表明,气溶
胶在远距离传输过程中,存在多次蒸发/凝结循环的云过程[16,18],与酸性组分反应的酸化过程[9,25]等,这些过程都会增加气溶胶中微量元素的溶解度.

表2 AS组与DS组气溶胶中微量元素溶解度(%)Table 2 Trace elements solubilities (%) both in AS and DS groups in Qingdao aerosols

图3 AS组与DS组气溶胶中微量元素的富集因子Fig.3 Enrichment factors of various trace elements in AS and DS groups in Qingdao aerosols
2.3.2 正矩阵因子分析 采用PMF方法对气溶胶的来源进行解析(模型为EPA PMF 3.0).结果显示2012年12月青岛气溶胶主要有7个来源:土壤源、二次生成源、机动车一次排放源、生物质燃烧源、燃煤排放源、海洋源和冶金工业源(各组分模型模拟值与实测值的相关系数均大于0.90,模型模拟结果可以反映各组分的真实信息)[26].由每个气溶胶样品中不同源相对贡献大小的定量解析结果可知,二次生成源对于 12月2、14~16、27和28日采集的样品贡献最大,海洋源对于3、13和31日采集的样品贡献最大,土壤源对4~5日和29日采集的样品贡献最大.比较这3类主要来自不同源的气溶胶样品中微量元素溶解度(图4),发现主要受土壤源影响的气溶胶中微量元素溶解度明显低于受二次生成源和海洋源影响的气溶胶;受二次生成源影响的气溶胶中Fe、Al、Pb、Ba、Cu、Mn和Zn的溶解度明显高于受海洋源影响的气溶胶, 而Bi、V、Cd、As的溶解度与海洋源气溶胶中的基本相当.海洋源和土壤源气溶胶中微量元素溶解度的差异,可能是由于海洋源气溶胶中粒子的比表面积较之土壤源气溶胶中的大 40%左右而更有利于微量元素溶解度提高.二次生成源和土壤源气溶胶中微量元素溶解度的差异,除了与气溶胶粒子的比表面积较之土壤源气溶胶中的大 60%左右有关外,还与其中高浓度的酸组分有关.二次生成源气溶胶中SO42-和NO3-等酸组分的浓度分别为142和174nmol/m3,远高于土壤源和海洋源中的约80和100nmol/m3,因此,大气酸化过程可能是二次生成源气溶胶中微量元素溶解度高的主要原因.

图4 不同来源的气溶胶中微量元素的溶解度Fig.4 Solubilities of various trace elements in aerosols originated from different sources
2.4 大气酸过程对气溶胶中微量元素溶解度的影响
气溶胶传输过程中与大气中酸性污染物混合发生的酸化反应可能是增加气溶胶中微量元素溶解度的重要原因之一[27].气溶胶中的酸性组分主要包括无机的SO42-和NO3-以及有机的二元
羧酸,其中乙二酸(C2O42-)是二元羧酸中最重要的组分.由于海盐粒子对气溶胶中的 SO42-也有一定的贡献,因此,为反映人为污染的影响,文中采用非海盐 SO42-(nss-SO42-)的浓度([nss-SO42-]=[SO42-]-0.2455×[Na+], 0.2455为海水中SO42-与Na+的质量比值).
以往的研究多关注硫化物的非均相转化反应对气溶胶中微量元素溶解度的影响[28],对 NOx的非均相转化过程的影响鲜有提及.本研究中溶解态微量元素与nss-SO42-、NO3-均显著相关,不仅表明了硫化物的非均相转化反应是控制气溶胶中微量元素溶解度的重要因子,而且暗示了NOx的大气化学过程可能也对其有重要贡献.另外,溶解态微量元素与 C2O42-也有显著相关关系,表明不仅无机酸组分、有机酸组分也可提高气溶胶中微量元素溶解度.

表3 青岛气溶胶中总的和溶解态的微量元素与酸性组分的相关关系Table 3 Correlation of total and soluble trace elements versus acid species in aerosols in Qingdao
2.5 天气条件对溶解度的影响
观测期间,青岛出现了雾、霾、雨/雪等特殊天气,利用中国气象局MICAPS天气图资料,将气溶胶样品按采样时的天气状况分类,其中,受雾天影响的样品4个,受降雨/雪影响的6个,受霾天影响的8个,另有13个未见以上特殊天气的记为晴天样品.
比较不同天气下青岛气溶胶中微量元素溶解度的差异,发现微量元素溶解度基本在雾天时最高,其次是雨/雪天,霾天和晴天时的最低(图5).雾、霾发生时,气溶胶中往往含有较高浓度的酸组分,但出乎意料的是霾天时微量元素溶解度明显低于雾天和雨/雪天的.比较不同天气下气溶胶中酸组分的浓度,发现霾天和雾天时nss-SO42-、 NO3-、C2O42-的浓度基本相当,分别约为 100、140、1.0,明显高于雨/雪天的85、85、0.7nmol/m3.因此酸组分含量高低并非造成不同天气下微量元素溶解度差异的唯一主控因子.
最近Shi等[24]在研究气溶胶中Fe溶解度时,发现气溶胶以“湿气溶胶”状态存在时间的长短是影响Fe溶解度的重要因素.大气颗粒物在云中只有以“湿气溶胶”存在时,气溶胶表层的水膜吸收的酸组分才能提高Fe的溶解度,且存在时间越长这种促进作用越明显.观测期间,雾天、雨/雪天、霾天和晴天时的平均相对湿度分别为82%、79%、58%和53%.可见,雾天时>80%的相对湿度使气溶胶以“湿气溶胶”的状态存在而更有利于其中的微量元素与酸性组分作用,而霾天
时相对湿度<60%,因此尽管气溶胶中酸组分的浓度较高,但微量元素溶解度却明显低于雾天时的样品.进一步分析气溶胶中微量元素溶解度与采样时相对湿度的关系,发现Fe、Al、Ba、Cu、V、Cd、Mn和Zn的溶解度与相对湿度之间均存在显著的相关关系(r>0.53,P<0.01).这些结果表明仅有高浓度酸组分的存在并不能提高微量元素的溶解度,只有在高的相对湿度下,这些酸组分才能促进微量元素溶解度的增大.

图5 不同天气条件下气溶胶中微量元素的溶解度Fig.5 Solubilities of trace elements in aerosols during different weather conditions
3 结论
3.1 青岛大气气溶胶中微量元素溶解度以 Al和Fe的最低,其平均溶解度<5%;其次为Pb、Ba和Bi,溶解度为10%左右;Cu、V、Cd和Mn的溶解度较高,约为20%~30%;Zn和As的最高,约为40%.
3.2 气溶胶中微量元素溶解度随沙尘负载量增大均呈规律性递减,且其与气溶胶粒子的比表面积有一定相关关系,表明微量元素溶解度与颗粒物的大小有关.
3.3 气溶胶的来源不同显著影响微量元素溶解度,受人为源影响的气溶胶中微量元素溶解度明显高于受沙尘源影响,造成这种差异的原因可能是气溶胶在传输中经历的大气过程,而非气溶胶中元素的初始来源或组成.
3.4 大气酸化过程是影响微量元素溶解度的主要因子.不仅 SO42-和 NO3-的非均相转化反应影响气溶胶中微量元素溶解度,有机酸对其也有一定贡献.
3.5 雾、霾天时,尽管气溶胶中均含有高浓度的酸组分,但霾天时微量元素溶解度明显低于雾天的,二者差异的主要原因是由于雾天时酸组分在较高的相对湿度下更能促进微量元素溶解度提高.
[1] Butler A. Acquisition and utilization of transition metal ions by marine organisms [J]. Science, 1998,281(5374):207-208.
[2] Mackie D S, Boyd P W, Mctainsh G H, et al. Biogeochemistry of iron in Australian dust: From eolian uplift to marine uptake [J]. Geochemistry Geophysics Geosystems, 2008,9(3):1-24.
[3] Falkowski P G, Barber R T, Smetacek V. Biogeochemical controls and feedbacks on ocean primary production [J]. Science, 1998,281(281):200-206.
[4] Morel F M M, Price N M. The biogeochemical cycles of trace metals in the oceans [J]. Science, 2003,300(5621):944-947.
[5] Paytan A, Mackey K R M, Chen Y, et al. Toxicity of atmospheric aerosols on marine phytoplankton [J]. Proceedings of the National Academy of Sciences of the United States of America, 2009, 106(12):4601-4605.
[6] Manousakas M, Papaefthymiou H, Eleftheriadis K, et al. Determination of water-soluble and insoluble elements in PM2.5by ICP-MS [J]. Science of the Total Environment, 2014, 493:694-700.
[7] Birmili W, Allen A G, Bary F, et al. Trace metal concentrations and water solubility in size-fractionated atmospheric particles and influence of road traffic [J]. Environmental Science & Technology, 2006,40(4):1144-1153.
[8] Fu H B, Shang G F, Lin J, et al. Fractional iron solubility of aerosol particles enhanced by biomass burning and ship emission in Shanghai, East China [J]. Science of the Total Environment, 2014,481(2):377–391.
[9] Hsu S C, Wong G T F, Gong G C, et al. Sources, solubility, and dry deposition of aerosol trace elements over the East China Sea [J]. Marine Chemistry, 2010,120(s1–4):116–127.
[10] Buck C S, Landing W M, Resing J A, et al. The solubility and deposition of aerosol Fe and other trace elements in the North Atlantic Ocean: Observations from the A16N CLIVAR/CO2repeat hydrography section [J]. Marine Chemistry, 2010,120(1):57-70.
[11] Buck C S, Landing W M, Resing J A. Pacific Ocean aerosols:Deposition and solubility of iron, aluminum, and other trace elements [J]. Marine Chemistry, 2013,157(12):117-130.
[12] Mahowald N M, Baker A R, Bergametti G, et al. Atmospheric global dust cycle and iron inputs to the ocean [J]. Global Biogeochemical Cycles, 2005,19(4):1064-1067.
[13] Baker A R, Croot P L. Atmospheric and marine controls on aerosol iron solubility in seawater [J]. Marine Chemistry, 2012, 120(1-4):4-13.
[14] Heal M R, Hibbs L R, Agius R M, et al. Total and water-soluble trace metal content of urban background PM10, PM2.5and black smoke in Edinburgh, UK [J]. Atmospheric Environment, 2005, 39(8):1417-1430.
[15] Baker A R, Jickells T D. Mineral particle size as a control on aerosol iron solubility [J]. Geophysical Research Letters, 2006, 33(17):254-269.
[16] Desboeufs K V, Losno R, Colin J L. Factors influencing aerosol solubility during cloud processes [J]. Atmospheric Environment, 2001,35(20):3529-3537.
[17] Desboeufs K V, Sofikitis A, Losno R, et al. Dissolution and solubility of trace metals from natural and anthropogenic aerosol particulate matter [J]. Chemosphere, 2005,58(2):195-203.
[18] Mackie D S, Boyd P W, Hunter K A, et al. Simulating the cloud processing of iron in Australian dust: pH and dust concentration [J]. Geophysical Research Letters, 2005,32(6):83-100.
[19] 贲孝宇,石金辉,仇 帅,等.青岛大气气溶胶中铁的溶解度及其影响因素 [J]. 环境科学学报, 2015,35(1):65-71.
[20] 石金辉,张 云,李瑞芃,等.东海大气气溶胶中无机氮组分的分布特征 [J]. 环境科学, 2010(12):2835-2843.
[21] Chan C, Yao X. Air pollution in mega cities in China [J]. Atmospheric Environment, 2008,42(1):1-42.
[22] Baker A R, Jickells T D, Witt M, et al. Trends in the solubility of iron, aluminium, manganese and phosphorus in aerosol collected over the Atlantic Ocean [J]. Marine Chemistry, 2006,98(1):43-58.
[23] Taylor S R. Trace element abundances and the chondritic Earth model [J]. Geochimica Et Cosmochimica Acta, 1964,28(12):1989-1998.
[24] Shi Z B, Woodhouse M T, Carslaw K S, et al. Minor effect of physical size sorting on iron solubility of transported mineral dust [J]. Atmospheric Chemistry & Physics, 2011,11(16):8459-8469.
[25] Sullivan R C, Guazzotti S A, Sodeman D A, et al. Direct observations of the atmospheric processing of Asian mineral dust [J]. Atmospheric Chemistry & Physics, 2006,7(5):1213-1236.
[26] 仇 帅,张爱滨,刘 明.青岛大气总悬浮颗粒物中微量元素的含量特征及其来源解析 [J]. 环境科学学报, 2015,35(6):1667-1675.
[27] Meskhidze N, Chameides W L, Nenes A, et al. Iron mobilization in mineral dust: Can anthropogenic SO2emissions affect ocean productivity? [J]. Geophysical Research Letters, 2003,30(21):267-283.
[28] Zhuang G, Yi Z, Duce R A, et al. Link between iron and sulphur cycles suggested by detection of Fe (n) in remote marine aerosols [J]. Nature, 1992,355:537-539.
Solubility of trace elements in atmospheric aerosols and determination factors in Qingdao, China.
ZHU Min2, SHI Jin-hui1,2*, BEN Xiao-yu3, QIU Shuai2, GAO Hui-wang1,2, YAO Xiao-hong1,2(1.Key Laboratory of Marine Environmental Science and Ecology, Ministry of Education, Ocean University of China, Qingdao 266100, China;2.College of Environmental Science and Engineering, Ocean University of China, Qingdao 266100, China;3.College of Physical and Environmental Oceanography, Ocean University of China, Qingdao 266100, China). China Environmental Science, 2016,36(11):3245~3252
Thirty-one 24-h total suspended particulate (TSP) samples were collected at an urban site in Qingdao in December 2012. The total concentrations of eleven trace elements in the samples were determined as well as their water-extracted concentrations. The solubility was <5% (on average, the same definition is applicable for latter) for Fe and Al, ~10% for Pb, Ba and Bi, 20%~30% for Cu, V, Cd and Mn; and ~40% for Zn and As. The determination factors of the solubility were further studied, in terms of dust loading, the origins of aerosol, acid processing and weather conditions. In general, the obtained solubility was increased with decreasing dust loadings. Combining the results from the backward trajectory clustering analysis and the positive matrix factorization (PMF), the solubility of trace elements in TSP mainly originated from anthropogenic sources was found to be larger than the value in TSP originated from Asian dust. The same was true when secondary and marine aerosols were compared against soil-derived aerosols. Atmospheric acidification processes can increase the solubility, depending on aging extents. SO42-and NO3-as well as the organic acids played a role in acidification processes. In addition, fog events apparently favored a higher solubility when comparing to haze events because of higher relative humidity therein.
trace elements;aerosol;solubility;acidification processes;haze
X513
A
1000-6923(2016)11-3245-08
朱 敏(1992-),女,山东济南人,中国海洋大学环境科学与工程学院硕士研究生,主要从事海洋大气环境化学研究.
2016-03-01
国家重大科学研究计划项目(2014CB9537002);国家自然科学基金资助项目(41210008;41176097)
* 责任作者, 教授, engroup@ouc.edu.cn
猜你喜欢
成都信息工程大学学报(2022年3期)2022-07-21
世界科学技术-中医药现代化(2021年5期)2021-11-05
辐射防护通讯(2019年3期)2019-04-26
试题与研究·中考化学(2016年4期)2017-03-28
中学化学(2016年10期)2017-01-07
中国光学(2015年5期)2015-12-09
中学化学(2015年5期)2015-07-13
国外科技新书评介(2014年4期)2014-12-17
