
O2分子在F掺杂锐钛矿型TiO2(001)面上吸附影响的研究*
2016-12-09 02:35刘华忠马为川
功能材料 2016年11期
刘华忠,马为川
(武汉东湖学院 基础课部, 武汉 430212)
O2分子在F掺杂锐钛矿型TiO2(001)面上吸附影响的研究*
刘华忠,马为川
(武汉东湖学院 基础课部, 武汉 430212)
利用DFT+U方法研究了氟离子掺杂对锐钛矿型TiO2(001)面对O2分析吸附的影响。共研究了由F离子分别取代表面上的2配位或3配位O离子,或表面上5配位Ti离子下的3配位O离子所形成的3种F离子杂质(分别记为FⅠ、FⅡ和FⅢ)的影响。F掺杂的表面与分子的作用结果表明,FⅠ杂质能够提高表面的稳定性,而FⅢ能提高表面的活性;由掺入FⅡ离子的表面与未掺杂的表面的活性相当。结果表明,3种形式F离子掺杂能显著提高的TiO2表面对O2分子的吸附能力。电子结构分析显示,F掺杂所形成的Ti3+在增强表面对分子的吸附能力中起着重要的作用。
TiO2;O2分子;吸附;第一性原理
0 引 言
TiO2作为一种廉价无毒、性能稳定的光催化材料,已被大量的理论与实践研究证明可以用于废水处理、空气净化等应用领域[1-3]。然而TiO2最主要的缺陷为其能带具有较宽的带隙(锐钛矿型为3.2 eV),这使得它只能利用太阳光中不足5%的能量[4]。要提高TiO2对太阳光的利用效率,一种有效手段就是在TiO2中掺非金属元素。据报道,通过在TiO2中掺入碳、氮、氟[5]、硫[6]或碘元素[7],都能不同程度地提高TiO2对可见光的响应能力。然而,与其它非金属元素掺杂的TiO2相比,氟元素掺杂由于在可见光区不仅对于提高TiO2光催化活性有着较大的潜力,而且能够提高TiO2在光催化过程中起着重要作用的高活性面的稳定性[8-9],目前正受到越来越多的关注。锐钛矿型TiO2(001)面由于具有较高的表面活性而在光催化应用中的重要作用已被广为证实[10-11]。然而由于锐钛矿型TiO2(001)面较高的表面能使其稳定性较差,这使得纯净的锐钛矿型TiO2(001)面在实际中的应用受到了限制。令人欣慰的是,实验和理论研究表面,氟离子掺杂可以使锐钛矿型TiO2(001)面的表面能下降到甚至低于钛矿型TiO2(101)面的表面能的水平,因而(001)面的稳定性可以得到显著提高[8-9]。此外,氟离子掺杂还能够提高TiO2晶粒的稳定性,能够防止晶粒彼此之间的粘连,还能阻止晶粒由锐钛矿相转变为催化活性较低的金红石相[12-13]。这些富有成效的研究揭示了氟离子掺杂在开发具有高活性(001)面并具有高稳定性的TiO2光催化剂颗粒方面的光明前景。
由于氟离子掺杂能够提高TiO2的稳定性以及光催化性能,人们在氟离子掺杂方面有着越来越多的兴趣。但氟离子在提高TiO2的光催化性能的作用机制仍然不太清楚,现有文献中的观点很多存在着争论甚至是相悖的结论[14]。到目前为止,关于含Ti3+的n型掺杂的TiO2表面,特别是F掺杂的锐钛矿型(001)面,对吸附在表面上的气体分子的影响的理论研究并不多见。因此,利用第一性原理方法探索气体分子与氟掺杂的TiO2表面的作用,将有助于进一步理解氟杂质在n型掺杂的TiO2在降解有害气体分子中的所起的作用。
1 计算方法与结构模型
本文的周期性密度泛函理论计算均采用开源软件包QUANTUM ESPRESSO中的PWscf模块进行的,采用基于广义梯度近似的GGA+U方法以及PW91交换-关联泛函的超软赝势方法。本文中所采用的U值为3.3 eV,该值已经被证明足以正确描述锐钛矿型TiO2中Ti原子的3d电子[15]。
纯净的TiO2(001)面采用一个(2×2)的表面超胞模拟,超胞的面积为0.758 nm×0.758 nm,厚度包含4个O—Ti—O亚层,约0.78 nm;超胞中一共包含12个原子层以及16个TiO2单元结构。将一个纯净的(2×2)的表面超胞中的一个O原子用F原子取代后,即得到氟掺杂的TiO2(001)表面片层模型。纯净的与氟掺杂的片层采用的Monkhorst-Pack网格k点密均为(2×2×1)。在所有的片层模型中,下面两个O—Ti—O亚层中的所有原子均被固定在体结构的位置上,而上面两个O—Ti—O亚层中的原子与表面上所吸附的分子均经过充分的弛豫。
2 F掺杂的锐钛矿(001)表面
在计算分子在氟掺杂的表面前,先需要了解氟掺杂对表面性质的影响。氟掺杂的方法为将表面上一个O原子替换为一个F原子。这里考虑3种替换方法,及超胞表面上的一个O2C原子被一个氟原子取代,或超胞表面上一个O3C原子被一个F原子替换,或位于表面的Ti5C原子之下的一个O3C原子被F原子取代分别记为FⅠ、FⅡ与FⅢ,相应的F掺杂的TiO2表面分别记为FⅠT,FⅡT以及FⅢT。这3种氟杂质均与Ti5C原子相邻,且具有不同的深度,分别代表这表面外层的氟杂质以及表面内部的氟杂质。与纯净表面上的O原子相比,F原子到邻近的原子之间的距离更大。例如,在FⅠT表面上(如图1(b)所示),两个非对称的F—Ti键的键长分别为0.2019与0.2248 nm,长于纯净表面上O2C原子两个近邻的Ti原子之间的距离(分别0.1747与0.2224 nm)。FⅡT表面上F-Ti6C的键长(如图1(c))与FⅢT表面上的F—Ti5C的键长(如图1(d))分别为0.2280与0.2126 nm,也都分别大于纯净表面上对应位置的键长(0.1983与0.1938 nm)。除此之外,F杂质没有给表面晶格带来很大的扭曲变形。FⅠT,FⅡT以及FⅢT这3种表面的表面能分别为0.74,0.92与1.04 J/m2,显示这3种F杂质对表面活性有不同的影响。

图1 纯净的与F掺杂的锐钛矿型TiO2(001)面的优化结构
从投影态密度(如图2所示)可以看出,对于纯净的TiO2片层,其费米能级位于带隙的中间位置;而当掺入F离子后,费米能级被推高到导带底部。在这3种F掺杂方式中,氟离子都会在带隙中略低于费米能级的位置引入定域化的自旋极化峰,这个峰即为Ti3+的能态。这个能态,是由于掺入F离子后,与原来未掺入F相比,片层内多了一个富余的未配对电子的结果。由极化理论[16],这个未配对电子会进入一个Ti4+的3d轨道,从而将其还原成Ti3+,停留在一个定域化的Ti3+轨道中。这可由图2中FT的投影态密度证实。 从PDOS分析中可由看出,在导带底部出现了一个高度定域化的峰,这个峰即对应着由F掺杂后引入的未配对电子形成的Ti3+。值得注意的是,在3种氟掺杂的片层中,F离子所产生的Ti3+均位于次表层与F离子近邻或次近邻的Ti6C原子上。处于Ti3+的3d轨道中的这个未配对电子,使体系具有了1 μB的总磁矩。这个未配对的电子引入了Ti3+这个还原态后,使体系变成了一个n型半导体,因而也就改变了表面的化学性质。关于F离子掺杂在体系中引入Ti3+这一结果已被相关的理论研究证实[14,17]。实验上,一个定域化的Ti3+图景能够更好地与TiO2输运过程中的电子跳跃机制相吻合[14,16]。定域的Ti3+中的富余电子也可能对表面的吸附性能带来影响。它们可能在改变表面的化学性质的过程中起着关键作用,甚至能完全改变表面的化学性质,譬如,能够给TiO2表面上O2的吸附与分解所必要的电子提供源泉[18]。
3 O2分子的吸附
催化剂表面上由于O2吸附产生的活性氧物种通常是表面催化反应中的重要参与物[19-20]。Anpo等[20]曾推测O2很容易吸附于n型半导体表面。因此,研究O2在氟掺杂的表面上吸附及其机制将对于理解F掺杂的TiO2表面的催化有积极的意义。通过将O2分子放在纯净的锐钛矿型TiO2(001)面上不同位置优化后可以发现,O2在这种表面上只有负吸附能,表明O2在这种表面上吸附是个吸热过程,因而吸附不能自然发生。这与以前的研究结论一致[21]。然而,当TiO2(001)面上掺入氟杂质后,发现O2可以较稳固的吸附上去,同时伴随着明显的电荷转移(见表1)。在这些氟掺杂的表面模型上(FⅠT,FⅡT与FⅢT),O2在表面上吸附的最稳定构型均通过两个O原子与同一个5配位Ti原子成键,分子中的O—O键平行于表面(如图3所示)。图3(a)~(c)分别显示了O2分子在FⅠ、FⅢ和FⅢ掺杂的表面的吸附构型;图3(d)~(f)显示了相应的电荷转移分布与总静电荷,其中棕红色区域表示得到电子,蓝色区域表示失去电子。在3种氟掺杂的表面上,O2在FⅢT上表面上吸附最强,FⅢ对于增强O2吸附的效应最明显,其吸附能为1.67 eV,为三者中最高(见表1所示)。

图2 纯净的与F掺杂的锐钛矿TiO2(001)表面的投影态密度

图3 O2分子在F掺杂的表面上最稳定的吸附构型及对应的电荷转移分布
Fig 3 Optimized geometries and charge transfer of an O2molecule on F-doped surfaces in the most stable configurations
投影态密度图给出了O2吸附的成键机制。如图4所示,导带底的Ti3+的定域态比较靠近费米能级,并位于O2的2π*反键轨道之上,这促使电子从Ti3+的3d轨道转移到O2的2π*反键轨道上。在此过程中,O2充当电子受主并使Ti3+消失,同时形成O2-超氧离子。作为吸附体系的磁矩的来源,2π*反键轨道中的3个电子使O2-超氧离子具有顺磁性。当电荷转移发生后,Ti3+也就消失了。Ti3+所对应的能态也就移动到费米能级之上,同时相应的Ti3d轨道变成空轨道(如图4所示)。
表1 O2分子在气态或以最稳定构型吸附在F掺杂的锐钛矿型TiO2(001)面时的结构参数(键长)、吸附能(Eads)、电荷转移数量(ΔQ)以及体系的总磁矩(M)
Table 1 Relaxed structural parameters (bond lengths), adsorption energies (Eads), charge transfer (ΔQ) and magnetic moments (M) of O2molecule in gas phase or adsorbed on F-doped surfaces in the most stable configurations
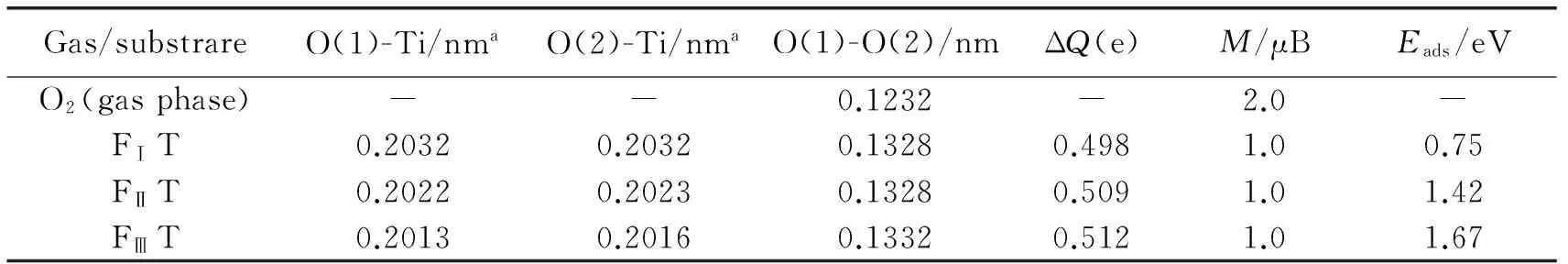
Gas/substrareO(1)-Ti/nmaO(2)-Ti/nmaO(1)-O(2)/nmΔQ(e)M/μBEads/eVO2(gasphase)--0.1232-2.0-FⅠT0.20320.20320.13280.4981.00.75FⅡT0.20220.20230.13280.5091.01.42FⅢT0.20130.20160.13320.5121.01.67
注:a为O原子的标识见图3;b为电荷转移的数量通过Bader电荷布局分析得到。
由于2π*反键轨道上增加了一个电子,被吸附的O2分子内的O—O键的键长明显增加。电荷的这种转移将费米能级移动到带隙中较低的位置,使体系变为一个p型半导体。此外还可以看出,由于FⅢ杂质更多的离子化特征,使得O2在FⅢT面上吸附得更强。这与带隙中源自于Ti3+的杂质态的出现对于O2的吸附有明显的促进作用相符。一般地,带隙中源自于Ti3+的杂质态对于O2的吸附有明显的增强效应,O2充当电子受主,O2吸附伴随着明显的电荷转移。在此过程中,Ti3+充当电子从表面转移到O2分子过程中必要的桥梁作用。这种电荷转移能减少表面中电子-空穴对的复合[22]。因而,TiO2的氟掺杂导致的表面这种电子结构与性质的改变,对有O2参与的表面催化的应用有重要的意义。
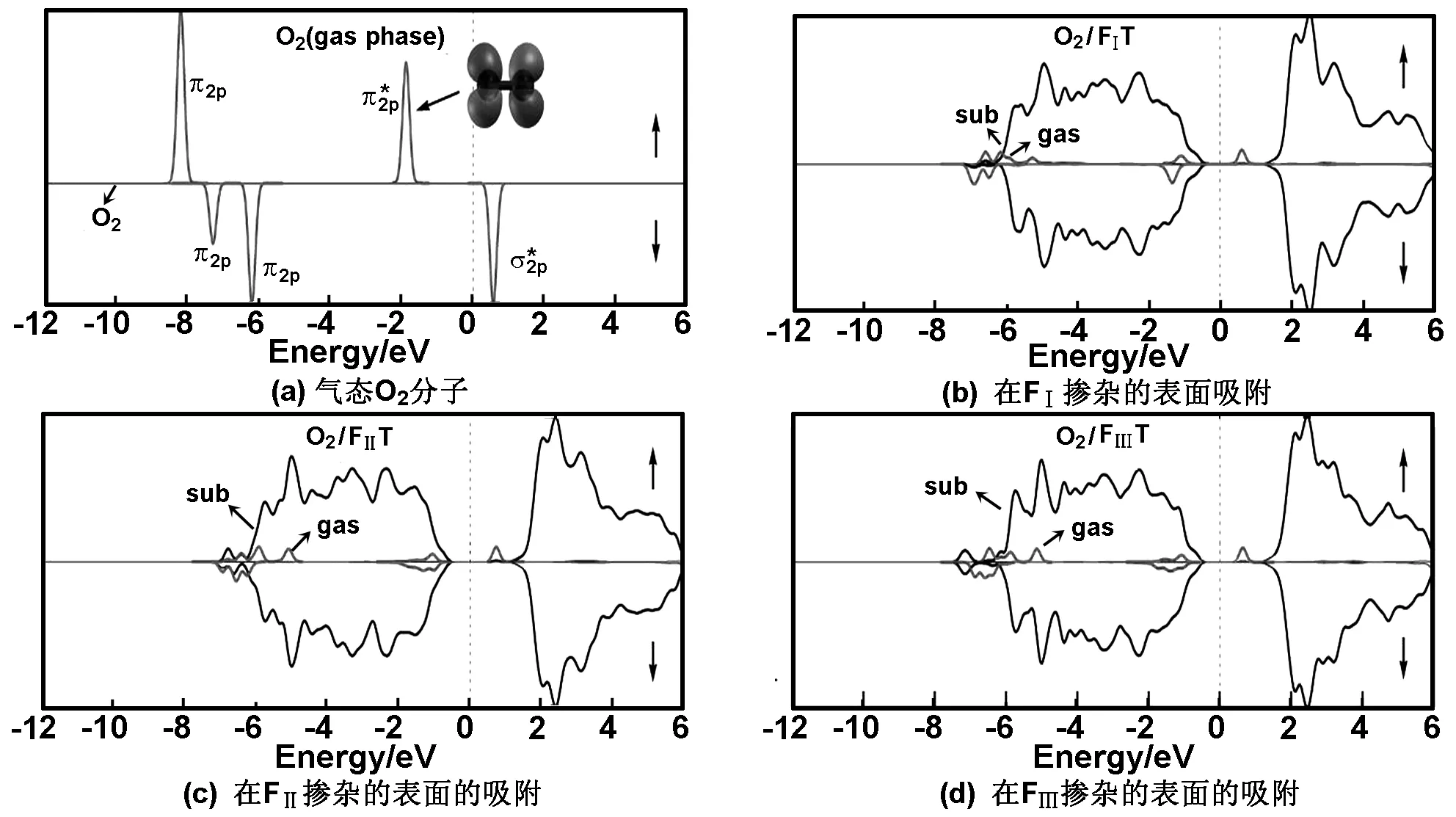
图4 气态O2分子的总态密度与O2分子吸附在表面时的投影态密度
Fig 4 Total density of states (TDOS) for a gas-phase O2molecule and PDOS for O2on FⅠT, O2on FⅡT and O2on FⅢT. The Fermi level is set to zero. The notation of “sub” denotes the states of substrate, and “gas” denotes the states of the adsorbed molecule (O2)
4 结 论
利用第一性原理讨论了O2分子在纯净的与F掺杂的锐钛矿型TiO2(001)面上最稳定的吸附构型及其电子结构等性质。计算表明,与在纯净表面上的吸附相比,F杂质引入的富余电子能提高电荷转移的数量,从而使吸附变得更强。特别是,在纯净的表面上O2的吸附并不能自发进行,而当表面里存在F杂质后,O2能够较强地吸附在表面上。在此过程中,Ti3+起着关键作用,这主要是Ti3+的定域化电子提高了费米能级,从而使电荷从表面转移到O2分子得以顺利进行。这些结果显示,体系中的Ti3+不仅对体系的电子结构性质和磁性质有着独特的影响,而且也对气体分子吸附后的结构性质也有影响。这些结果对于更好地理解F杂质在提高TiO2表面活性与稳定性方面提供重要的解释角度。F掺杂引入的Ti3+在电荷转移与强化气体分子吸附中的桥梁作用显示了这个物种在TiO2基光催化剂的未来应用中的具有重要的应用潜力。
[1] Fujishima A, Honda K. Electrochemical photolysis of water at a semiconductor electrode[J].Nature, 1972, 238(5358): 37-38.
[2] Hoffmann M R, Martin S T, Choi W,et al. Environmental applications of semiconductor photocatalysis[J].Chemical Reviews, 1995, 95(1): 69-96.
[3] Khan S U M, Al-Shahry M, Ingler W B. Efficient photochemical water splitting by a chemically modified n-TiO2[J]. Science, 2002, 297(5590): 2243-2245.
[4] Chen X, Mao S S. Titanium dioxide nanomaterials: synthesis, properties, modifications, and applications[J].Chemical Reviews, 2007, 107(7): 2891-2959.
[5] Yue Yuanxia, Feng Qing, Wang Yin. Defect formation energy and electronic properties of anatase TiO2doped with C, N, F[J]. Journal of Functional Materials, 2013, 44(13): 1879-1883.
[6] Liu Jianmei, Zhu Zhongqi, Zhang Jin, et al. Research of S-doped modified TiO2photocatalyst[J]. Journal of Functional Materials, 2014, (1): 6-9.
[7] Wang Chuntao, Zhang Wenkui, Huang Hui,et al. Preparation of I-doped TiO2film electrode and photoelectrochemical properties[J]. Journal of Functional Materials, 2011, (8): 1445-1447.
[8] Yang H G,Sun C H,Qiao S Z,et al. Anatase TiO2single crystals with a large percentage of reactive facets[J]. Nature, 2008, 453(7195): 638-641.
[9] Han X, Kuang Q, Jin M,et al. Synthesis of titania nanosheets with a high percentage of exposed (001) facets and related photocatalytic properties[J]. Journal of the American Chemical Society, 2009, 131(9): 3152-3153.
[10] Vittadini A, Selloni A, Rotzinger F P,et al. Structure and energetics of water adsorbed at TiO2anatase (101) and (001) surfaces[J]. Physical Review Letters, 1998, 81(14): 2954-2957.
[11] Diebold U, Ruzycki N, Herman G S,et al. One step towards bridging the materials gap: surface studies of TiO2anatase[J]. Catalysis Today, 2003, 85(2-4): 93-100.
[12] Xu J, Ao Y, Fu D,et al. Low-temperature preparation of F-doped TiO2film and its photocatalytic activity under solar light[J]. Applied Surface Science, 2008, 254(10): 3033-3038.
[13] Yang H, Zhang X. Synthesis, characterization and computational simulation of visible-light irradiated fluorine-doped titanium oxide thin films[J].Journal of Materials Chemistry, 2009, 19(37): 6907-6914.
[14] Di Valentin C, Pacchioni G, Selloni A. Reduced and n-type doped TiO2: nature of Ti3+species[J]. The Journal of Physical Chemistry C, 2009, 113(48): 20543-20552.
[15] Mattioli G, Filippone F, Alippi P,et al. Ab initio study of the electronic states induced by oxygen vacancies in rutile and anatase TiO2[J]. Physical Review B, 2008, 78(24): 241201.
[16] Austin I G,Mott N F. Polarons in crystalline and non-crystalline materials[J]. Advances in Physics, 2001, 50(7): 757-812.
[17] Czoska A M, Livraghi S, Chiesa M,et al. The nature of defects in fluorine-doped TiO2[J]. The Journal of Physical Chemistry C, 2008, 112(24): 8951-8956.
[18] Wendt S, Sprunger P T, Lira E,et al. The role of interstitial sites in the Ti3d defect state in the band gap of titania[J]. Science, 2008, 320(5884): 1755-1759.
[19] Che M, Eley D D,Paul B W. Tench in characterization and reactivity of molecular oxygen species on oxide surfaces, Vol. Volume 32[M]. Academic Press, 1983:1-148.
[20] Anpo M, Che M, Fubini B,et al. Generation of superoxide ions at oxide surfaces[J].Topics in Catalysis, 1999, 8(3-4): 189-198.
[21] Hussain A, Gracia J, Nieuwenhuys B E,et al. Chemistry of O- and H-containing species on the (001) surface of anatase TiO2: a DFT dtudy[J]. Chemphyschem, 2010, 11(11): 2375-2382.
[22] Yu J C, Yu Ho, Jiang Zhang. Effects of F-doping on the photocatalytic activity and microstructures of nanocrystalline TiO2powders[J]. Chemistry of Materials, 2002, 14(9): 3808-3816.
O2molecules in F doped anatase TiO2(001) to study the adsorption effect on the surface
LIU Huazhong,MA Weichuan
(Basic Courses department, Wuhan Donghu University,Wuhan 430212,China)
Using the DFT+U method, we investigated the influence of F-dopants on the activities of TiO2anatase (001) surface. Three kinds of F-dopants including FⅠ, FⅡand FⅢ, which are the F-dopants substituting a surface O2Catom, or a surface O3Catom, or an O3Catom below a surface Ti5Catom, were considered. The results of interaction between F-doped surfaces and molecules show that FⅠdopant raises the stability of surface, while FⅢdopant raises the activity of surface. The surface doped with FⅡhas an activity similar to the pristine one. The results show that the adsorption of O2molecule can be strengthened significantly by the three kinds of F-dopants. The electronic structure analysis shows that Ti3+induced by F-dopants plays an important role in enhancing interaction between gas molecules and TiO2surfaces.
TiO2; O2molecule; adsorption; the first-principles
1001-9731(2016)11-11110-05
2015-12-10
2016-03-10 通讯作者:刘华忠,E-mail: hzliew@163.com
刘华忠 (1979-),男,湖北大悟人,讲师,博士,主要从事光催化材料研究。
O647.32
A
10.3969/j.issn.1001-9731.2016.11.022
猜你喜欢
四川地质学报(2022年2期)2022-07-08
矿产勘查(2020年8期)2020-12-25
河北科技大学学报(2020年3期)2020-07-14
广州化工(2020年5期)2020-03-31
原子与分子物理学报(2020年5期)2020-03-17
陶瓷学报(2019年5期)2019-01-12
无机盐工业(2017年6期)2017-03-11
中国资源综合利用(2016年9期)2016-01-22
应用化工(2014年11期)2014-08-16
应用化工(2014年7期)2014-08-09
