
气相中催化CO与N2O循环反应的密度泛函理论研究
2016-12-06 01:31:07王永成张玉伟王晓莉
西北师范大学学报(自然科学版) 2016年6期
王永成,张玉伟,王晓莉,李 爽,盛 阳
(西北师范大学化学化工学院,甘肃兰州 730070)

王永成,张玉伟,王晓莉,李 爽,盛 阳
(西北师范大学化学化工学院,甘肃兰州 730070)

密度泛函理论(DFT);转化频率(fTO);能量跨度模型;反应机理
近来,如何降低大气污染物一直是科学家们关注的焦点[1-2].作为大气污染物之一的N2O和CO倍受理论化学研究者的关注,N2O不仅会引起温室效应,还会破坏臭氧层,引起臭氧空洞;CO也是一种有毒气体,它可以结合血液中的血红蛋白并阻止与氧结合从而导致血液缺氧症.大量研究表明,过渡金属氧化物离子作为催化剂之所以能减少N2O和CO的污染[3-4],是由于过渡金属氧化物离子由于具有较高的选择性和活性,因此过渡金属氧化物被广泛用作催化剂和起催化作用的介质材料[5-7].氧化物的催化作用主要是提供氧,在实验中,许多过渡金属氧化物离子与一些小分子CO,C2H4,C2H2等[8-10]之间的氧转移反应已经被观察到,其反应在热力学和动力学上是有利的.同时,过渡金属氧化物转移氧之后的产物也能在碰撞条件下与N2O继续反应.鉴于它们在氧转移催化反应中表现出优良的催化活性,过渡金属氧化物催化剂的研究依然是具有挑战性的课题[11].

(1)
(2)
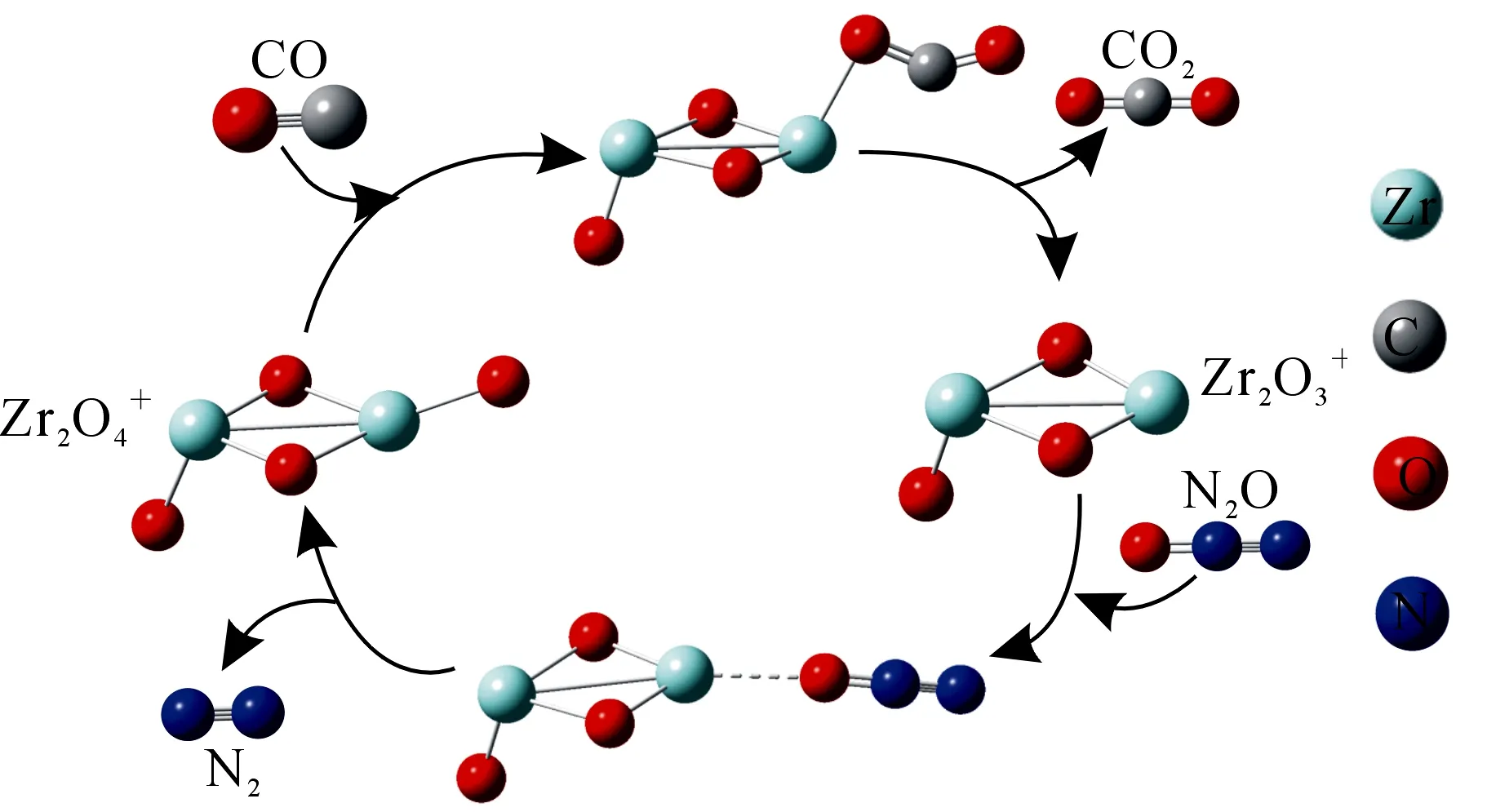
图催化CO与N2O的循环反应示意图
1 计算方法和理论背景
1.1 全参数优化几何构型
在理论化学计算中密度泛函理论[13](DFT)已被广泛运用.文中采用DFT中Becke’s三参数交换泛函(B3)结合Lee-Yang-Parr(LYP)相关泛函的混合DFT/Hartree-Fock的B3LYP方法[14-15],对C,H,O,N采用TZVP[16-17]全电子基组,对Zr原子选用有效核心势(ECP)LANL2DZ赝势基组[18].全参数优化了二、四重态反应势能面上所有驻点的几何构型,以及对优化后的稳定点做了频率分析,保证稳定构型的力常数均大于零,过渡态鞍点处有唯一虚频.为了确保各过渡态及反应路径的合理性,对各过渡态鞍点进行了内禀反应坐标(IRC)[19]验证.二、四重态反应势能面上各驻点构型见图2,势能面相对能量见图3.所有计算均采用Gaussian09程序包完成[20-21].
1.2 能量跨度模型
在实验中,催化性能的评价是通过测定单位时间内单位浓度催化剂的转换次数,即催化转化频率TOF.Kozuch通过结合催化转化频率的概念和Eyring速率常数公式,建立起由Gibbs自由能描述催化循环过程的能量跨度模型[22-23].在能量跨度模型理论中,并不是每个中间体和过渡态的Gibbs自由能对整个循环反应速率都有影响,而起决定作用的是Gibbs自由能最低的中间体和Gibbs自由能最高的过渡态,即整个循环反应的决速中间体(TDI)和决速过渡态(TDTS).Kozuch又结合Compbell对循环反应速率控制度的定义,提出了循环反应控制度的概念[24-25],进而计算各中间体和过渡态的控制度.
2 结果与讨论

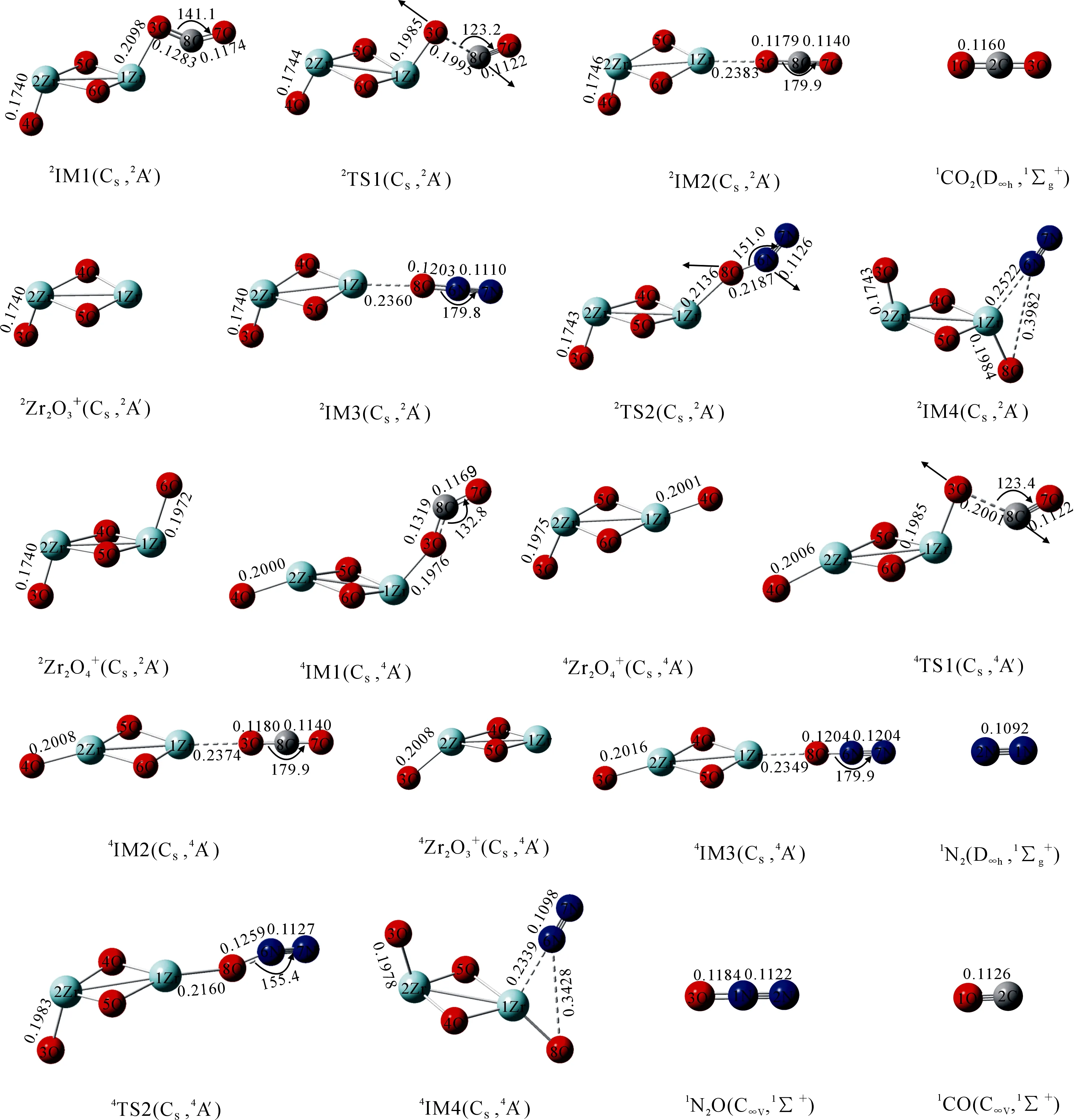
图2 在B3LYP/TZVP水平下各驻点优化后的几何构型及相关参数(键角/(°),键长/nm)
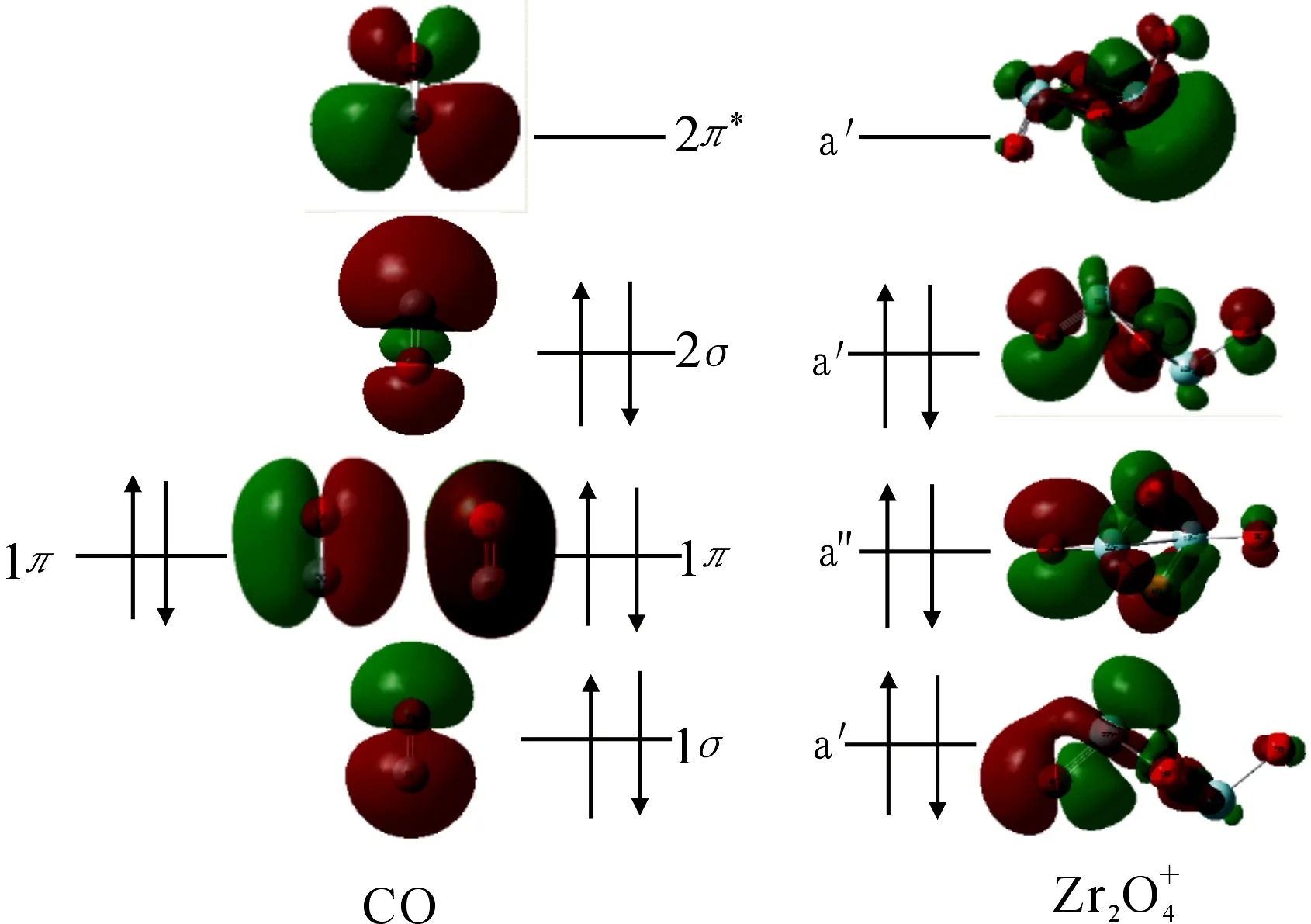
图3 初始反应物的前线分子轨道相互作用分析图
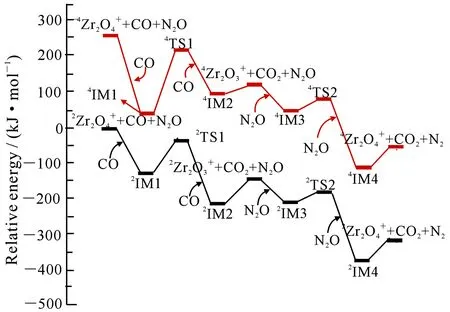
图4 在二重态和四重态下所有反应的路径示意图

2.2 循环反应中催化剂的TOF计算
根据Kozuch能量跨度模型理论,在循环反应中对反应速率起决定作用的是Gibbs自由能最低的决速中间体(TDI)和Gibbs自由能最高的决速过渡态(TDTS),而不是活化能最大的基元决速步骤.能量跨度δE作为多步催化反应的表观活化能,它的大小由TDI和TDTS间的Gibbs自由能差值决定.

图催化CO与N2O循环反应的相对Gibbs自由能图

物种IM1IM2IM3IM4XTOF,TiTS1TS2XTOF,Ij180×1072118×10216142×10-209575×10146531×1026455×10-135645×10147595×1027509×10-134126×10281117×10161100100923×10-121100
(3a)
(3b)
(4)
其中,ΔGr为最终产物和初始反应物的Gibbs自由能的差值;Ti为第i个过渡态的Gibbs自由能值;Ij为第j个中间体的Gibbs自由能值.
3 结论
1)整个反应是沿着二重态势能面进行,是典型的单态反应;
2)该催化循环反应是一个强放热反应;
[1] PLANE J M C,ROLLASON R J.A kinetic study of the reactions of Fe(a5D)and Fe+(a6D) with N2O over the temperature range 294-850 K[J].JChemSoc,FaradayTrans,1996,92(22):4371.
[2] RITTER D,WEISSHAAR J C.Kinetics of neutral transition-metal atoms in the gas phase:oxidation of titanium(a3F) by nitric oxide,oxygen,and nitrous oxide[J].JPhysChem,1989,93(4):1576.
[3] CAMPBELL M L,KÖLSCHl E J,HOOPER K L.Kinetic study of the reactions of gas-phase V(a4F3/2),Cr(a7S3),Co(a4F9/2),Ni(a3F4,a3D3) and Zn(4s2 1S0) atoms with nitrous oxide[J].JPhysChemA,2000,104(47):11147.
[4] TISHCHENKO O,CEULEMANS A,NGUYEN M T.Theoretical study on the group 2 atoms+ N2O reactions[J].JPhysChemA,2005,109(27):6099.
[5] TRIMM D L.HandbookofHeterogeneousCatalysis[M].Weinheim:Wiley-VCH,1997.
[6] TROVARELLI A.Catalytic properties of ceria and CeO2-containing materials[J].CatalRevSciEng,1996,38(4):439.
[7] BARTEAU M A.Organic reactions at well-defined oxide surfaces[J].ChemRev,1996,96(4):1413.
[8] ZEMSKI K A,JUSTES D R,CASTLEMAN A W.Reactions of group V transition metal oxide cluster ions with ethane and ethylene[J].JPhysChemA,2001,105(45):10237.

[10] JOHNSON G E,MITRIC R,TYO E C,et al.Stoichiometric zirconium oxide cations as potential building blocks for cluster assembled catalysts[J].JAmeChemSoc,2008,130(42):13912.
[11] CRAMER C J,TRUHLAR D G.Density functional theory for transition metals and transition metal chemistry[J].PhysChemChemPhys,2009,11:10757.
[12] JOHNSON G E,MITRIC R,TYO E C,et al.Stoichiometric zirconium oxide cations as potential building blocks for cluster assembled catalysts[J].JAmChemSoc,2008,130(42):13912.
[13] SU M D,CHU S Y.Density functional study of some germylene insertion reactions[J].JAmChemSoc,1999,121(17):4229.
[14] BECKE A D.Density-functional thermochemistry.Ⅲ.The role of exact exchange[J].JChemPhys,1993,98:5648.
[15] STEPHENS P J,DEVIN F J,CHABALOWSKI C F,et al.Ab initio calculation of vibrational absorption and circular dichroism spectra using density functional force fields[J].JPhysChem,1994,98(45):11623.
[16] ANDREA D,HAEUSSERMANN U,DOLG M,et al.Energy-adjusted ab initio pseudopotentials for the second and third row transition elements[J].TheorChimActa,1990,77(2):123.
[17] GILB S,WEIS P,FURCHE F,et al.Structures of small gold cluster cations(Au+n,n<14):ion mobility measurements versus density functional calculations[J].JChemPhys,2002,116:4094.
[18] HAY P J,WADT W R.Ab initio effective core potentials for molecular calculations.Potentials for the transition metal atoms Sc to Hg[J].JChemPhys,1985,82(1):270.
[19] GONZALEZ C,SCHLEGEL H B.Reaction path following in mass-weighted internal coordinates [J].JChemPhys,1990,94(14):5523.
[20] KOZUCH S,SHAIK S.How to conceptualize catalytic cycles?The energetic span model[J].AccChemRes,2011,44(2):101.
[21] FORESMAN J B,ORTIZ J V,CIOSLOWSK J,et al.Gaussian 09,Revision D.01[Z].Wallingford:Gaussian Inc,2009.
[22] CHRISTIANSEN J A.The elucidation of reaction mechanisms by the method of intermediates in quasi-stationary concentrations[J].AdvCatal,1953(5):311.
[23] KOZUCH S,SHAIK S.A combined kinetic-quantum mechanical model for assessment of catalytic cycles:application to cross-coupling and heck reactions[J].JAmChemSoc,2006,128(10):3355.
[24] STEGELMANN C,ANDREASEN A,CAMPBELL C T.Degree of rate control:how much the energies of intermediates and transition states control rates[J].JAmChemSoc,2009,131(23):8077.
[25] YOSHIZAWA K,SHIOTA Y,YAMABE T.Intrinsic reaction coordinate analysis of the conversion of methane to methanol by an iron-oxo species:a study of crossing seams of potential energy surfaces[J].JPhysChem,1999,111(2):538.
(责任编辑 陆泉芳)
WANG Yong-cheng,ZHANG Yu-wei,WANG Xiao-li,LI Shuang,SHENG Yang
(College of Chemistry and Chemical Engineering,Northwest Normal University,Lanzhou 730070,Gansu,China )
density functional theory(DFT);turnover frequency(fTO);energy span model;reaction mechanism.
10.16783/j.cnki.nwnuz.2016.06.013
2016-05-12;修改稿收到日期:2016-07-14
国家自然科学基金资助项目(21263023)
王永成(1956—),男,陕西户县人,教授,博士研究生导师.主要研究方向为化学动力学.
E-mail:ycwang@163.com
O 641
A
1001-988Ⅹ(2016)06-0064-06
猜你喜欢
建材发展导向(2022年14期)2022-08-19 02:10:52
北京航空航天大学学报(2022年5期)2022-06-06 09:27:18
中国药学药品知识仓库(2022年10期)2022-05-29 02:59:32
大学化学(2021年8期)2021-09-26 10:51:16
西部交通科技(2021年9期)2021-01-11 18:28:15
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
上海建材(2018年4期)2018-11-13 01:08:54
电脑知识与技术(2018年3期)2018-03-21 09:27:04
哈尔滨理工大学学报(2017年1期)2017-04-08 04:16:24
