
高效液相-电喷雾串联质谱法测定动物肌肉组织中的糖皮质激素
2016-12-01 08:50:13龚兰陈明魏娴邹春王冉
分析化学 2016年1期
龚 兰 陈 明 魏 娴 邹 春 王 冉
(江苏省农业科学院食品质量安全与检测研究所,农业部农产品质量安全控制技术与标准重点实验室,
高效液相-电喷雾串联质谱法测定动物肌肉组织中的糖皮质激素
龚 兰 陈 明 魏 娴 邹 春 王 冉*
(江苏省农业科学院食品质量安全与检测研究所,农业部农产品质量安全控制技术与标准重点实验室,
省部共建国家重点实验室培育基地-江苏省食品质量安全重点实验室,南京 210014)
本实验建立了肌肉组织中氢化可的松、可的松、泼尼松和地塞米松含量的高效液相色谱-电喷雾串联质谱(LC-ESI-MS/MS)检测方法。样品经酶水解、乙酸乙酯提取、HLB固相萃取净化,C8色谱柱分离,在多反应选择性监测模式(MRM)下采用负离子模式进行信号采集和测定。4种糖皮质激素的检出限为0.13~0.25 μg/kg,定量限为0.25~0.5 μg/kg。在0.5~50.0 μg/L范围内具有良好的线性关系(R2>0.99)。在0.5, 5.0和10.0 μg/kg基质加标水平下,4种物质的平均回收率为74.0%~101.8%,相对标准偏差0.7%~8.6%。
糖皮质激素; 高效液相-电喷雾串联质谱; 固相萃取; 肌肉组织
1 引 言
糖皮质激素是一种固醇类激素,在临床上对人体有明显的消炎作用,也可起到免疫抑制和抗过敏作用[1,2]。最早被人们所认知的糖皮质激素有可的松和氢化可的松,属于内源性糖皮质激素,之后又合成了疗效更好的人工糖皮质激素,临床常用的合成糖皮质激素有地塞米松、泼尼松和泼尼松龙等,图1为常见4种糖皮质激素结构图[3~6]。
在动物饲养过程中,糖皮质激素还可起到生长促进作用,因此部分饲养者违规使用糖皮质激素类药物[7,8]。不恰当的使用可导致动物源性食品中药物残留,影响食用者健康,如内分泌紊乱和消化系统疾病等[9]。因此,许多国家已经对动物源食品中合成糖皮质激素残留量进行明确、严格的管理和规定。我国农业部在235号公告明确指出地塞米松在猪、牛、马的肌肉和肾脏中不得超过0.75 μg/kg,肝脏中不得高于2 μg/kg,奶中含量不得超过0.3 μg/kg[10],但没有对可的松和氢化可的松进行限量规定。
目前,有许多分析技术应用于检测分析糖皮质激素含量。放射免疫技术具有迅速、灵敏和高通量的特点,但在常规筛查中缺乏选择性[11]。高效液相色谱和气相色谱无法对样品进行定性分析,灵敏度和特异性都无法满足痕量兽药残留分析[12]。气相色谱串联质谱(GC-MS)虽然灵敏度高,选择性和特异性好,但是衍生过程繁琐,不易操作[13]。液相色谱串联质谱(LC-MS/MS)是分析动物源性食品中痕量兽药残留的重要方法,能够满足对低浓度样品的准确分析[14,15]。
由于肌肉样品基质比较复杂,国内对不同肌肉组织中糖皮质激素含量测定的相关报道较少。文献[4]指出部分糖皮质激素可与组织蛋白形成共轭化合物,影响提取效率。因此,提取样品中游离态和结合态的目标物,去除杂质,减少基质干扰是本实验的主要目的。本研究将对不同的肌肉组织进行酶解处理结合态目标物,固相萃取法净化浓缩样品,经LC-MS/MS检测,建立肌肉组织中氢化可的松、可的松、泼尼松和地塞米松4种糖皮质激素含量的检测方法,为肌肉中测定糖皮质激素含量提供参考。
2 实验部分
2.1 仪器、试剂与材料
液相色谱串联质谱(LC-MS/MS)联用仪(美国Agilent公司)由Agilent 1200和Agilent 6410三重四级杆串联质谱构成,配有电喷雾离子源(ESI); 氮气吹干仪(美国Organomation),MilliQ去离子水发生器(美国Millipore公司); 24位固相萃取装置(美国Supleco公司); 高速离心机(德国Eppendorf公司); 恒温振荡培养箱(太仓市强乐实验设备有限公司)。
图1 4种糖皮质激素类药物结构图Fig.1 Structural formulas of the investigated glucocorticoids
甲醇、甲酸(色谱纯,美国Honeywell公司); 乙酸乙酯、乙酸铵、(分析纯,国药公司); 超纯水(电阻率≥18.2 MΩ·cm);β-葡萄糖醛苷酶/芳基硫酸酯酶(>100000 units/mL,上海安谱公司); Oasis HLB固相萃取小柱(500 mg/6 mL,美国Waters公司)。
标准品可的松(Cortisone,COR)、氢化可的松(Hydrocortisone,HCOR)、地塞米松(Dexamethasone,DXM)(德国Dr Ehrenstorfer GmbH公司,纯度>97%); 泼尼松(Predisone,PNS,加拿大TRC公司,纯度98%)。
2.2 标准品配制
分别称取适量标准品,用甲醇配成1 mg/mL的标准贮备液,-20℃避光储存。准确移取上述标准储备液,以初始流动相稀释得到所需标准工作液,4℃储存备用。
2.3 样品前处理
(1)提取 肌肉组织均质后,准确称取2 g(精确至0.01 g)组织试样于50 mL离心管中,加入40 μLβ-葡萄糖醛苷酶/芳基硫酸酯酶和8 mL 0.2 mol/L乙酸铵缓冲液(pH 5.2),涡旋混匀,于40℃下避光振荡6 h。酶解后放置至室温,涡旋后10000 r/min离心5 min,取上层有机相,移入另一离心管中,加入10 mL 乙酸乙酯,涡旋混匀,5000 r/min离心5 min; 再将上层有机相移入另一离心管,下层加入10 mL乙酸乙酯,涡旋振荡,5000 r/min离心5 min,合并上层有机相,50℃氮吹尽干,以5 mL 30%(V/V)甲醇复溶样品。
(2)净化 Oasis HLB固相萃取柱依次用5 mL水、5 mL甲醇活化,样液全部过柱,再依次加入5 mL水、5 mL甲醇(2∶8,V/V)和5 mL正己烷淋洗、抽干,5 mL甲醇洗脱、收集并抽干,50℃氮气吹干,1.0 mL 30%乙腈-水(V/V)复溶,过0.22 μm滤膜,供液相色谱-质谱仪测定。
2.4 液相色谱条件
色谱柱:Agilent ZORBAX RX-C8(150 mm×2.1 mm,5 μm)进行分离,流动相A为0.1%甲酸,流动相B为乙腈,流速为0.3 mL/min,柱温30℃,进样量20 μL; 分离梯度洗脱程序为: 0~2 min,30%~40% B; 2~7 min,30%~59% B; 7.01 min,30% B。
2.5 质谱条件
ESI电喷雾离子源,负离子扫描模式(ESI),选择监测扫描模式(MRM),喷雾器压力为15 Psi,干燥气体流速10 L/min、离子源温度300℃,毛细管电压4000 V。各物质离子对优化后参数见表1。
2.6 方法验证
将混合标准工作液用初始流动相逐级稀释成0.5, 1.0, 2.0, 5.0,10.0, 20.0和50.0 μg/L系列标准工作液。称取2 g空白样品,经过2.3(1)节提取后,加入不同浓度标准品溶液复溶,配成系列浓度的基质混合标准工作液,采用外标法进行定量与定性分析,获得4种化合物的标准曲线、线性相关系数、方法检出限(LOD)和定量限(LOQ)。
取空白样品,添加3种不同浓度(0.5, 5.0和10 μg/kg)标准溶液,每个浓度水平平行测定6次,按照2.3(2)节进行提取净化,计算回收率和相对标准偏差(RSD),确定和计算方法的精密度和准确度。
表1 目标化合物的多离子反应监测及质谱参数
Table 1 Mass spectrometric parameters for the target compounds in MRM transition
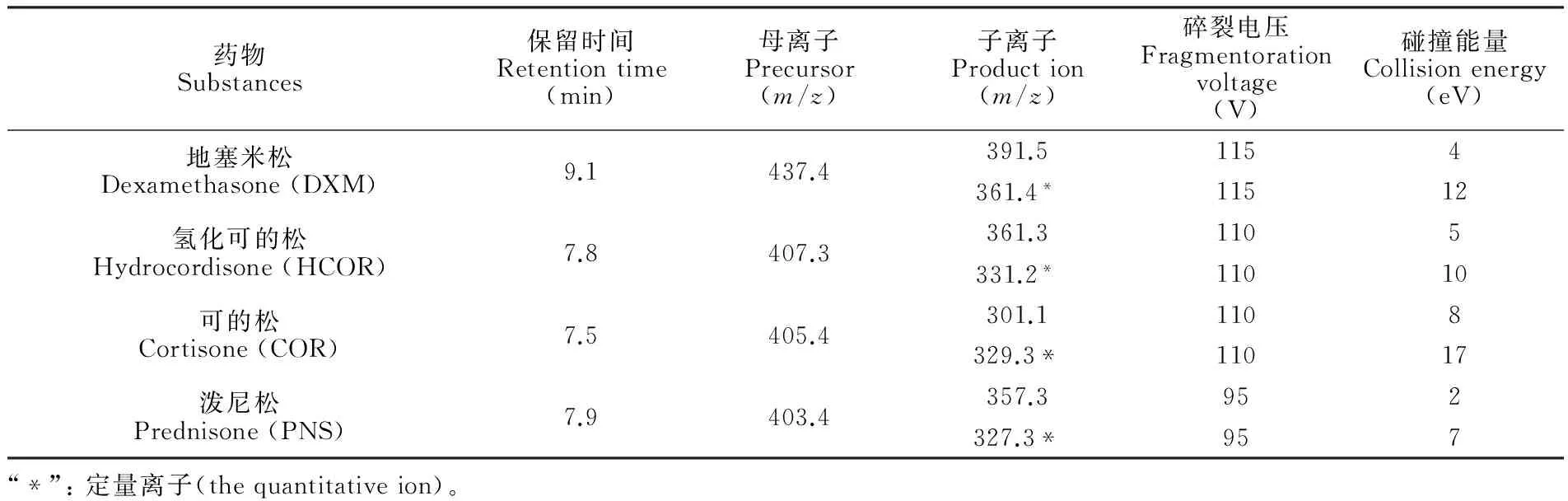
药物Substances保留时间Retentiontime(min)母离子Precursor(m/z)子离子Production(m/z)碎裂电压Fragmentorationvoltage(V)碰撞能量Collisionenergy(eV)地塞米松Dexamethasone(DXM)9.1437.4391.51154361.4*11512氢化可的松Hydrocordisone(HCOR)7.8407.3361.31105331.2*11010可的松Cortisone(COR)7.5405.4301.11108329.3*11017泼尼松Prednisone(PNS)7.9403.4357.3952327.3*957“*”:定量离子(thequantitativeion)。
3 结果与讨论
3.1 质谱条件的优化
分别在正负离子扫描模式下对糖皮质激素进行扫描,结果表明, ESI-扫描模式下的电喷雾离子化能够提供更多的碎片信息,同时灵敏度大幅提高。本实验以甲酸水作为流动相,在ESI-模式下通过全扫描模式发现, [M+HCOO]-丰度含量最高,这是由于目标物与甲酸形成共轭离子[M+HCOO]-,鉴于此选择[M+HCOO]-作为母离子,与文献[16]一致。在碰撞诱导解离过程中, 母离子在C21羟甲基的位置发生断裂,形成特征碎片离子[M-H-CH2O]-,进一步诱导解离可以失去H2O, CH4, HF和HCl等分子,产生特征子离子,可进行定量分析[17](图2)。
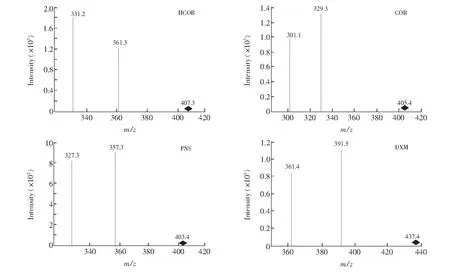
图2 4种糖皮质激素的[M+HCOO]-精确质量数提取质谱图Fig.2 Exaction product ion spectra of 4 glucocorticoidsHCOR: Hydrocortisone; COR: Cortisone; PNS: Predisione; DXM: Dexamethasone.
3.2 液相条件的优化
为了确定最优的液相分离条件,选取不同规格的色谱柱(Agilent SB-C18,150 mm ×2.1 mm,3.5 μm; Agilent XDB-C18,150 mm ×2.1 mm,3.5 μm; Agilent Rx-C8,150 mm ×2.1 mm,5.0 μm和Agilent SB-C18,150 mm ×4.6 mm,5.0 μm),反相色谱柱C18和C8均可以分离目标物,其中Agilent Rx-C8(150 mm ×2.1 mm,5.0 μm)色谱柱分离效果良好,出峰时间较为合适。
糖皮质激素类药物化学结构相似,因此在分离上存在一定的难度。采用甲醇体系时,各化合物无法完全分离,此时共流出的杂质较多,对信号干扰增强,响应值较低[13]。乙腈不仅能将二者分离,响应值较高,而且可以得到较完美的峰形。甲酸是糖皮质激素电离诱导过程不可缺少的物质,实验发现,甲酸含量过低,会影响目标物与甲酸离子的结合; 甲酸浓度较高,影响目标物离子化效率,选择0.1%甲酸较为合适(图3)。实验发现,pH值和温度对液相分离没有明显影响,与文献[18]一致。
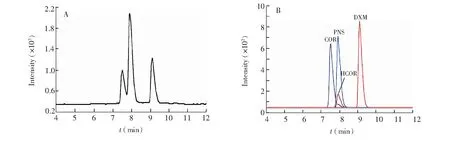
图3 50 ng/mL的4种糖皮质激素类药物的分离色谱图(A)和MRM质谱图(B)Fig.3 Separated chromatograms (A) and mutiple reaction monitoring (MRM ) chromatograms (B) of 4 glucocorticoids at 50 ng/mL
3.3 样品提取
从文献[2,19]中得知糖皮质激素在动物体内的存在方式有游离态和结合态的共轭态化合物,因此提高提取效率和获得较准确的数据首先需要将这类结合态的激素从动物组织中释放出来,通常采用水解方式进行。本实验中碱水解采用1 mol/mL NaOH 8 mL,时间6 h、温度40℃; 酸水解8 mL 1 mol/mL HCl,时间6 h、温度40℃; 酶水解选取β-葡萄糖醛苷酶/芳基硫酸酯酶,温度40℃、pH 5.2,时间6 h。取2 g阳性肉样,按照上述方案进行水解,得出酸水解和酶解的提取率明显高于碱水解,但酸水解不利于后续药物的提取操作,因此选择酶水解,这与文献[16]一致。
糖皮质激素属弱极性化合物,易溶于有机试剂,实验考察了3种提取试剂:甲醇、乙酸乙酯和叔丁基甲醚。3种物质提取效率见表2,甲醇提取后溶液浑浊,样液不利于后续SPE净化,且提取杂质较多; 乙酸乙酯萃取效率为86.0%~92.3%,经质谱测定,目标峰无其它基质干扰,灵敏度高; 叔丁基甲醚作提取溶剂时,4种化合物的回收率均低于50%。本研究选乙酸乙酯作为提取剂。
表2 3种有机试剂萃取效率对比表
Table 2 Extraction efficiency of 3 kinds of organic reagent
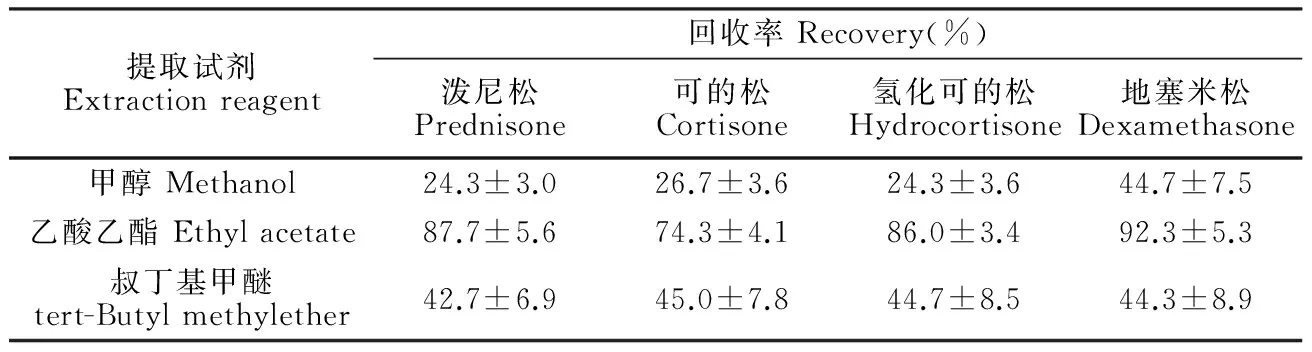
提取试剂Extractionreagent回收率Recovery(%)泼尼松Prednisone可的松Cortisone氢化可的松Hydrocortisone地塞米松Dexamethasone甲醇Methanol24.3±3.026.7±3.624.3±3.644.7±7.5乙酸乙酯Ethylacetate87.7±5.674.3±4.186.0±3.492.3±5.3叔丁基甲醚tert-Butylmethylether42.7±6.945.0±7.844.7±8.544.3±8.9
实验表明,不同浓度甲醇溶液复溶提取后的干物质,影响后续HLB固相萃取小柱净化效率。考察了体积分数为10%, 30%, 50%和70%甲醇对小柱回收率的影响(图4),其回收率最高为30%(V/V)甲醇,因为高浓度甲醇与HLB载体竞争,将弱极性目标药物洗脱下来,而30%甲醇不仅使目标物保留在柱体,而且能较好移除部分杂质。
3.4 样品净化-固相萃取
SPE固相萃取可以减少直接提取带来的基质干扰,实验考察了4种小柱(Waters Oasis HLB 500 mg,6 mL; 月旭 C18500 mg,6 mL; SUPELCLEAN LC-18 500 mg,3 mL; Waters Silica 500 mg,6 mL)对回收率的影响,小柱装载50 μg/L 目标化合物,按照萃取柱推荐方法进行,回收率结果(图5)表明,泼尼松、可的松和氢化可的松回收率最高的是Oasis HLB,平均回收率为87.3%~90.0%,对4种物质有较好的保留;其次是LC-18和Silica,并且发现C18小柱无法保全部药物,Silica小柱也有部分药物损失,导致3种糖皮质激素回收率低。4种固相萃取小柱对地塞米松的提取净化效率都高于80%,但最理想的是Oasis HLB,平均回收率达到105.6%。综上所述,采用HLB可以实现4种物质同时提取,并达到较高提取率。

图4 不同浓度甲醇水复溶后提取效果对比图Fig.4 Comparing of extraction efficienties in different concentrations of methanol-water(V/V)10%CH3OH-H2O; 30%CH3OH-H2O; 50%CH3OH-H2O; 70%CH3OH-H2O.
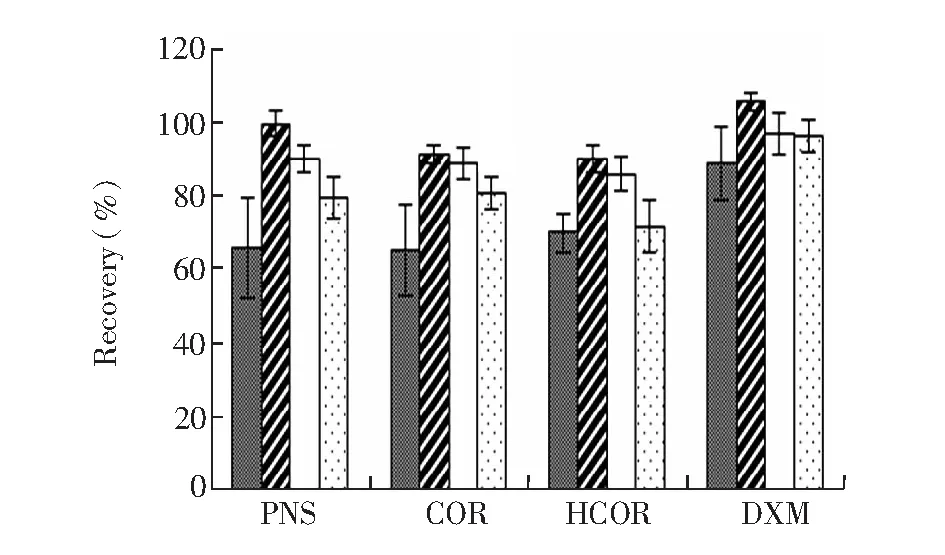
图5 糖皮质激素在4种固相萃取小柱中的回收率Fig.5 Recoveries of 4 glucocorticoids with different SPEC18; HLB; LC-18; Silica.
洗脱液选取甲醇、乙酸乙酯和正己烷-丙酮(6∶4,V/V)进行对比,实验发现,乙酸乙酯和正己烷-丙酮无法将所有目标物有效地从小柱上洗脱下来,回收率低于60%,甲醇洗脱回收率高于70%,鉴于此,选择甲醇作为最佳的洗脱液。
3.5 线性范围和检出限、定量限
检出限LOD定义为空白样品3倍信噪比时的浓度; 定量限LOQ定义为空白样品10倍信噪比时的浓度[18]。根据此要求得到4种物质的LOD和LOQ(表3)。选取空白样品配制成0.5, 1.0, 2.0, 5.0, 10.0, 20.0和50.0 μg/L 的7个系列的标准工作液,以目标物的峰面积Y对其含量X(μg/L)进行回归分析,其相关系数(R2)均大于0.99,表明各种糖皮质激素在0.5~50.0 μg/L范围内具有良好的线性关系(表3)。
表3 4种糖皮质激素的线性回归方程和线性范围
Table 3 Linear ranges and regression equations of 4 glucocorticoids

化合物Substances相关系数Correlationcoefficient(R2)线性范围Linearrange(μg/L)LOD(μg/kg)LOQ(μg/kg)泼尼松PNS0.99950.5~50.00.130.25可的松COR0.99920.5~50.00.130.25氢化可的松HCOR0.99651.0~50.00.250.50地塞米松DXM0.99830.5~50.00.130.25
3.6 方法回收率与精密度
选取猪肉、牛肉、鸡肉样品作为空白基质,添加水平为0.5, 5.0和10.0 μg/kg。回收率实验结果(表4)表明: 3种不同基质的平均回收率为74.0%~101.8%,RSD低于8.6%,符合实验要求。
3.7 实际样品分析
利用本方法测定了15份市售肉样(牛肉、猪肉和鸡肉各5份),部分牛肉、猪肉检出可的松和氢化可的松,但目前相关公告没有界定不同组织中内源和外源氢化可的松和可的松的含量。1份牛肉样品检出地塞米松超标,含量为5.44 μg/kg(表5)。5份鸡肉样品中均未检出糖皮质激素药物。
4 结 论
糖皮质激素类药物经过酶解作用,将结合态药物从肌肉中解离,乙酸乙酯提取样液,经HLB小柱净化去除杂质,通过LC-MS/MS分离检测,样品基质干扰小。本方法的灵敏度高,稳定性好,能够同时检测肌肉组织中内源和外源糖皮质激素。
表4 牛肉、猪肉和鸡肉中4种糖皮质激素的加标回收率及精密度(n=6)
Table 4 Spike recoveries and precision for 4 glucocorticoids in beef,pork and chicken(n=6)

基质Material化合物Substance0.5μg/kg回收率Recovery(%)RSD(%)5.0μg/kg回收率Recovery(%)RSD(%)10.0μg/kg回收率Recovery(%)RSD(%)牛肉Beef猪肉Pork鸡肉Chicken泼尼松PNS87.24.980.08.583.01.4可的松COR92.85.189.98.683.33.7氢化可的松HCOR88.63.997.75.498.47.0地塞米松DXM101.86.895.44.888.52.1泼尼松PNS79.24.374.03.576.50.7可的松COR85.13.991.94.097.12.7氢化可的松HCOR88.22.898.24.9101.72.1地塞米松DXM98.46.277.78.377.74.0泼尼松PNS81.95.984.53.582.35.9可的松COR87.94.896.97.276.24.4氢化可的松HCOR93.85.296.21.879.77.1地塞米松DXM100.82.675.04.084.56.3
表5 样品的测定结果
Table 5 Detection results of real samples

样品Sample样品编号Samplecode检测结果Found(μg/kg)地塞米松DXM泼尼松PNS可的松COR氢化可的松HCOR牛肉Beef猪肉Pork1NDNDND1.422NDNDND1.393NDND1.252.4945.44NDND1.805NDNDNDND1NDND1.002.882NDNDNDND3NDNDNDND4NDND0.3611.555NDNDND2.06ND:未检出(Notdetected)。
1 CUI Xiao-Liang, SHAO Bing, ZHAO Rong, TU Xiao-Ming.ChineseJournalofChromatography, 2006, 24(3): 213-217
崔晓亮, 邵 兵, 赵 榕, 涂晓明. 色谱, 2006, 24(3): 213-217
2 Deceuninck Y, Bichon E, Monteau F, Dervilly G P, Antignac J P, Bizec B L.J.Chromatogr.A, 2013, 1294: 76-86
3 SUN Xue-Ting, SHANG Shao-Ming, CHEN Xiu-Ying, WANG Yun, LI Juan.ChineseJ.Anal.Chem., 2014, 42(1): 36-40
孙雪婷, 商少明 , 陈秀英, 汪 云, 李 娟. 分析化学, 2014, 42(1): 36-40
4 Cuzzola A, Mazzini F, Petri A.J.Phar.Biomed.Anal., 2014, 94: 203-209
5 De Clercq N, Julie V B, Croubels S, Delahaut P, Vamhaecke L.J.Chromatogr.A, 2013, 1301(2): 111-121
6 LI Fei, GU Li-Li, ZHANG Yan, ZHAO Bao-Hua.FoodScience, 2010, 31(2): 154-156
李 飞, 谷丽丽, 张 岩, 赵宝华. 食品科学, 2010, 31(2): 154-156
7 Cherlet M, de Baere S, de Backer P.J.Chromatogr.B, 2004, 805(1): 57-65
8 Courtheyn D, Le Bizec B, Brambilla G, De Brabander H F, Cobbaert E, van de Wiele M, Vercammen J, de Wasch K.Anal.Chim.Acta., 2002, 473(1): 71-82
9 NIU Jin-Yang, SHI Hong-Xia.FoodScience, 2010, 31: 212-214
牛晋阳, 时宏霞. 食品科学, 2010, 31: 212-214
10 Ministry of Agriculture.No. 235BulletinoftheMinistryofAgricultureofthePeople′sRepublicofChina. [2008-06-29]
农业部. 中华人民共和国农业部公告第235号. [2008-06-29]
http://yz.hz-agri.gov.cn/uploadFiles/2005-10/1130221564406.doc
11 Difrancesco R, Frerichs V, Donnelly J, Hagler C, Hochreiter J, Tornatore K M.J.Chromatogr.B., 2007, 859(1): 42-51
12 XU Jin-Zhong, ZHANG Xiao-Yan, DING Tao, WU Bin, SHEN Chong-Yu, JIANG Tuan, LIU Fei.ChineseJ.Anal.Chem., 2009, 37(3): 341-346
徐锦忠, 张晓燕, 丁 涛, 吴 斌, 沈崇钰, 蒋 原, 刘 飞. 分析化学, 2009, 37(3): 341-346
13 YAN Hua, YUN Huan, LIU Xin, CUI Feng-Yun, LI Jian-Hui, GAO Yang-Yang, ZHANG Bin, DING Shuang-Yang, ZHANG Chao-Hui.JournalofInstrumentalAnalysis, 2013, 32(8): 909-914
严 华, 云 环, 刘 鑫, 崔凤云, 李建辉, 高洋洋, 张 杉, 丁双阳, 张朝辉. 分析测试学报, 2013, 32(8): 909-914
14 Herrero P, Borrull F, Pocurull E, Marcé R M.J.Chromatogr.A, 2012, 1224: 19-26
15 Callejas S L, Biddlecombe R A, Jones A E, Joyce K B, Pereira A I, Pleasance S.J.Chromatogr.B, 1998, 718: 243-250
16 van den Hauwe O, Dumoulin F, Antignac J P, Bouche M P, Elliott C, van Peteghem C.Anal.Chim.Acta, 2002, 473: 127-134
17 Cherlet M, de Baere S, de Backer P, Marcé R M.J.Chromatogr.A, 2004, 805: 57-65
18 Herrero P, Borrull F, Pocurull E.J.Chromatogr.A, 2012, 1224: 19-26
19 van den Hauwe O, Dumoulin F, Elliott C, Peteghem C V.J.Chromatogr.B, 2005, 817: 215-223
(Received 15 July 2015; accepted 14 October 2015)
This work was support by the National Natural Science Foundation of China (No. 31302009)
Determination of Glucocorticoids in Muscle Tissues by Liquid Chromatography-Electrospray Tandem Mass Spectrometry
GONG Lan, CHEN Ming, WEI Xian, ZOU Chun, WANG Ran*
(InstituteofFoodQualityandSafety,JiangsuAcademyofAgriculturalSciences,KeyLaboratoryofControlTechnologyandStandardforAgro-productSafetyandQualityMinistryofAgriculture,JiangsuKeyLaboratoryofFoodQualityandSafety-StateKeyLaboratoryCultivationBaseofMOST,Nanjing210014,China)
A method was developed to determine cortisone, cortisol, prednisone and dexamethasone in muscle tissues by high performance liquid chromatography-tandem mass spectrometry (LC-ESI-MS/MS). Deconjugation of glucocorticoids was carried out by enzymatic hydrolysis. These preliminary steps were followed by ethyl acetate extraction and HLB solid phase extraction clean up step for all matrices. Chromatographic separation was achieved on C8column and MS/MS data were obtained in the multiple reaction monitoring mode using negative electrospray ionization. Calibration graphs were prepared in the 0.5-50.0 μg/L range and good linearity was achieved (R2≥0.99).The limits of detection (LOQ) and the limits of quantification (LOD) were 0.13-0.25 μg/kg and 0.25-0.50 μg/kg, respectively. Spiking at the levels of 0.5, 5.0 and 10.0 μg/L, the average recovery for glucocorticoids ranged from 74.0% to 101.8%. The relative standard deviations (RSD) were between 0.7%-8.6%.
Glucocorticoids; High performance liquid chromatography tandem mass spectrometry; Solid-phase extraction; Muscle tissues
10.11895/j.issn.0253-3820.150561
本文系国家自然科学基金资助项目(No.31302009)
2015-07-15收稿; 2015-10-14接受
* E-mail: wangran2001@126.com
猜你喜欢
少年文艺(2022年8期)2022-07-08 10:02:47
中华养生保健(2020年1期)2020-11-16 00:47:44
中国经济周刊(2017年6期)2017-03-21 00:59:27
读写算·高年级(2016年3期)2016-05-30 01:53:46
橡胶工业(2015年2期)2015-07-29 08:29:46
中国医疗美容(2015年1期)2015-07-12 10:06:18
中国药业(2014年24期)2014-05-26 09:00:14
西南军医(2014年5期)2014-04-25 07:42:49
中国医学科学院学报(2014年6期)2014-03-11 20:26:18
食品工业科技(2014年9期)2014-03-11 18:15:39
