
超高效液相色谱法测定虾青素*
2016-11-30 08:13陈伟珠张怡评晋文慧方华洪专
化学分析计量 2016年6期
陈伟珠,张怡评,晋文慧,方华,洪专
(国家海洋局第三海洋研究所,国家海洋局海洋生物资源综合利用中心,福建省海洋生物资源开发利用协同创新中心,福建厦门 361005)
超高效液相色谱法测定虾青素*
陈伟珠,张怡评,晋文慧,方华,洪专
(国家海洋局第三海洋研究所,国家海洋局海洋生物资源综合利用中心,福建省海洋生物资源开发利用协同创新中心,福建厦门 361005)
建立超高效液相色谱法快速检测虾青素的方法。采用UPLC BEH C8色谱柱(50 mm ×2.1 mm,1.7 μm),考察了流动相、流量及柱温对虾青素样品分离的影响,确定了最佳色谱条件:等度洗脱,流动相为甲醇–水(体积比为75∶25),流量为0.5 mL/min,柱温为40℃,检测波长为475 nm。虾青素的质量浓度在0.2~10.0 µg/mL范围内与其色谱峰面积呈良好的线性关系,线性相关系数r=0.998 8,检出限(S/N=3)为0.1 µg/mL,定量限(S/N=10)为0.2 µg/mL。测定结果的相对标准偏差为0.41%(n=6),加标回收率为105.8%~110.3%。该方法快速、简单、可靠、灵敏、重复性好,可用于虾青素有关样品的快速检测。
虾青素;超高效液相色谱法;快速测定;杂质分离
虾青素(又名虾黄素、虾黄质)是虾蟹外壳、牡蛎、鲑鱼及某些藻类中含有的类胡萝卜素含氧衍生物,是一种酮式类胡萝卜素。经动物和临床实验研究表明,虾青素具有较强的抗氧化、抗癌抑癌、增强免疫、预防心血管疾病等保健功能[1],尤其抗氧化性比维生素E强100多倍,被称为“超级维生素E”。虾青素在食品添加剂、保健食品、医药、水产养殖和化妆品等方面有广泛的应用[2–3],美国等国家已允许其作为食品添加剂,市场前景广阔[4]。
目前,国内外对虾青素的检测方法主要有分光光度法[5–6]、高效液相色谱法[7–9]、高效液相色谱–质谱法[10–11]、超高效合相色谱法[12]和超高效液相色谱法[13]。但是文献方法只是针对虾青素含量的检测,而虾青素由于其分子结构存在共轭双键链及其末端的不饱和酮基和羟基,性质不稳定,在储藏过程中易与光、热、氧化物发生作用,产生其它杂质[4]。笔者对色谱柱、流动相、柱温和流量等色谱条件等进行优化,对虾青素及其杂质进行分离,建立了超高效液相色谱测定虾青素的方法,该方法简单、快速、准确,可用于虾青素及其有关杂质的检测。
1 实验部分
1.1 主要仪器与试剂
超高压液相色谱仪:Acuity型,附带自动进样器、柱温箱、TUV 检测器在线脱气机和Epower 2 色谱工作站,美国Waters公司;
Milii-Q 超纯水纯化系统:美国Millipore公司;
分析天平:AL104型,感量为0.1 mg,德国梅特勒公司;
2,6-二叔丁基对甲酚(BHT):分析纯,国药集团化学试剂有限公司;
虾青素标准品:纯度不小于97%,美国Sigma公司;
甲醇、乙腈:色谱纯,德国Merck公司;实验用水为超纯水。
1.2 色谱条件
色谱柱:ACQUITY UPLCTM C8色谱柱(2.1 mm×50 mm,1.7 μm,美国Waters公司);柱温:40℃;检测器:可调波长紫外检测器;检测波长:475 nm;流动相:甲醇–水(体积比为75∶25);流量:0.5 mL/min;进样体积:10 μL。
1.3 虾青素标准储备液配制
称取虾青素对照品1 mg,用乙腈溶解,置于10 mL 容量瓶中,稀释至标线,摇匀,配成质量浓度为100 μg/mL的虾青素标准储备液。
2 结果与讨论
2.1 色谱条件
2.1.1 色谱柱的选择
虾青素结构中含有一个长的共扼不饱和双键,在反相色谱柱上保留性较强。首先考察不同反相色谱柱对虾青素及其有关杂质分离效果的影响,分别选择UPLC BEH C18、UPLC BEH C8和UPLC CSH C18三种常见的UPLC色谱柱进行考察,色谱图见图1。图1结果表明,在甲醇–水(体积比为80∶20)和柱温30℃的相同条件下,虾青素在UPLC BEH C8色谱柱上不仅有保留,而且主峰与杂质峰均能分开;但在UPLC BEH C18和UPLC CSH C18色谱柱中的保留太强,虾青素在UPLC BEH C18色谱柱中的保留时间为9.34 min,在UPLC CSH C18中的保留时间更晚,12 min未出峰。因此选择UPLC BEH C8作为分析虾青素的色谱柱。
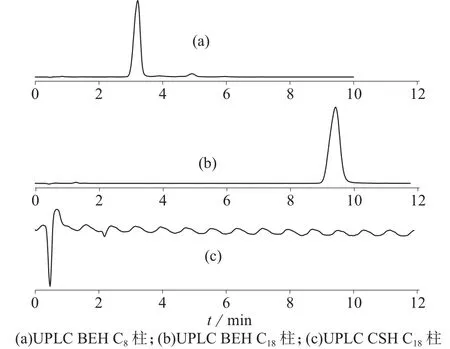
图1 高温破坏的虾青素在不同色谱柱上的色谱图
2.1.2 流动相的选择
比较了相同比例的乙腈–水和甲醇–水两种不同流动相对虾青素及其杂质分离效果的影响。结果表明,其它条件相同时,甲醇–水和乙腈–水作为流动相时,主峰均能与杂质峰有效地分离,但甲醇–水做流动相的分离效果更好,杂质峰之间也能基线分离,因此选择甲醇–水体系作为流动相。考察不同比例的甲醇–水对虾青素与其杂质峰分离效果的影响。在柱温30℃和流速0.3 mL/min 的相同条件下,改变甲醇–水的比例(75∶25,80∶20,85∶15,90∶10),不同比例流动相的色谱图如图2所示。
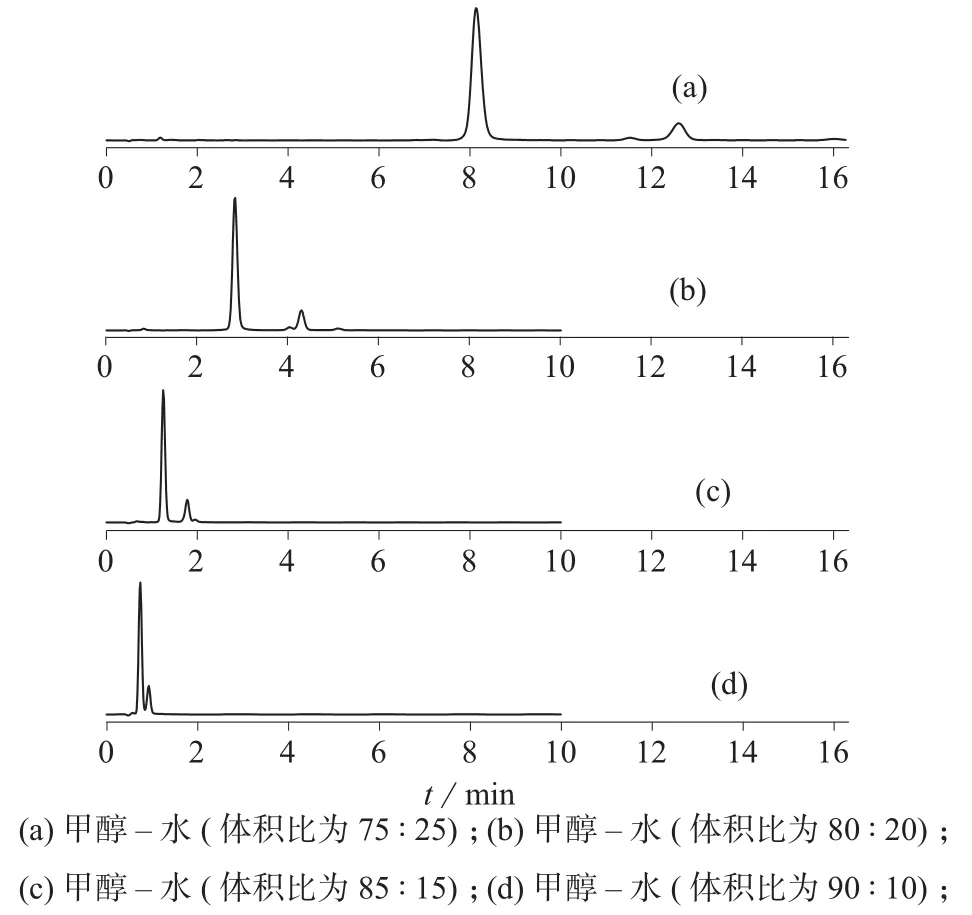
图2 高温破坏的虾青素在不同流动相条件下的色谱图
图2结果表明,在甲醇–水(体积比为75∶25)、甲醇–水(体积比为80∶20)和甲醇–水(体积比为85∶15) 3种流动相条件下,虾青素均能与其杂质峰有效地分离,但在甲醇–水(体积比为75∶25)作为流动相时,不仅虾青素能跟其它杂质分离,且杂质峰之间的分离效果更好;而当甲醇体积比增加到90%时,虾青素与杂质峰未能分离。因此,选择甲醇–水(体积比为75∶25)作为流动相。
2.1.3 柱温的选择
在流动相为甲醇–水(体积比为75∶25)和流量为0.3 mL/min 的相同条件下,考察柱温变化(30,35,40℃)对分离效果的影响,结果见图3。图3结果表明,在3种不同柱温条件下,虽然样品的保留时间随着柱温的提高而缩短,但变化不是特别明显,且样品主峰跟其它杂质峰均分离良好。因此考虑样品的保留时间,柱温选择40℃。
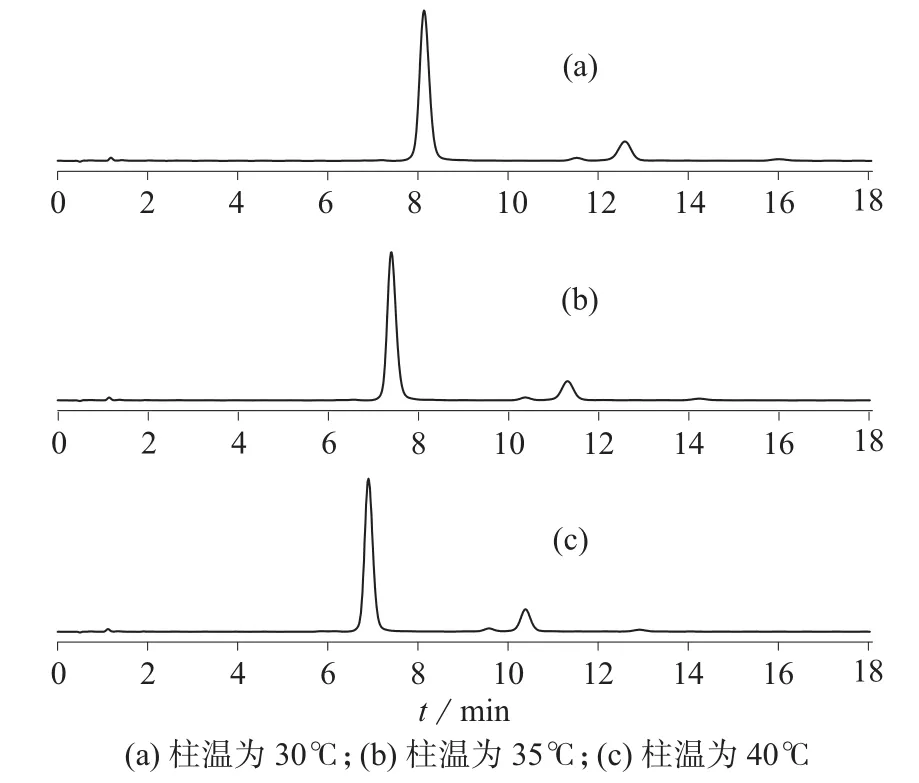
图3 高温破坏的虾青素在不同柱温条件下的色谱图
2.1.4 流动相流量的选择
在流动相为甲醇–水(体积比为75∶25)和柱温40℃的相同条件下,改变流动相流量(0.3,0.4,0.5,0.6 mL/min),考察流量对分离效果的影响,结果见图4。由图4可知,样品的保留时间随着流量的增加而缩短,分离度基本没有改变,但当流量增加到0.6 mL/min时,杂质峰之间未能很好地分开。因此流量选择0.5 mL/min。
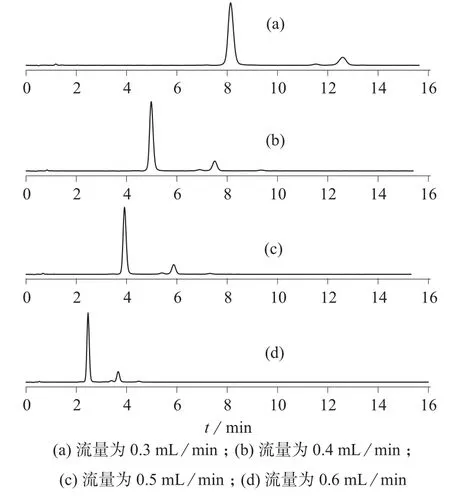
图4 高温破坏后的虾青素在不同流量下的色谱图
2.2 检出限与定量限
将对照品溶液逐级稀释后,以信噪比S/N=3 时的质量浓度为检出限,S/N=10 时的质量浓度为定量下限。计算得检出限为0.1 µg/mL,定量限为0.2 µg/mL。
2.3 线性关系
精密量取不同体积的虾青素标准储备液,配制质量浓度分别为0.2,0.4,1.0,2.0,4.0,8.0,10.0 μg/mL的系列标准工作溶液,按1.2色谱条件测定其峰面积,以峰面积(Y)对虾青素的质量浓度(X)进行线性回归。结果表明,虾青素质量浓度在0.2~10.0 μg/mL范围内线性关系良好,线性回归方程为Y=15 078X+3 365.7,相关系数r=0.998 8。
2.4 精密度试验
对质量浓度为8.0 μg/mL 的虾青素标准溶液连续进样测定6次,结果见表1。由表1可知,测定结果的相对标准偏差为1.42%,表明该方法具有良好的精密度。

表1 精密度试验结果
2.5 回收试验
精密称取不同量的虾青素,按照1.3制备供试品溶液,在相当于供试品溶液80%,100%,120%的浓度下,分别考察加标回收率,结果见表2。由表2可知,方法的回收率为105.8%~110.3%,表明该方法具有较高的准确度。
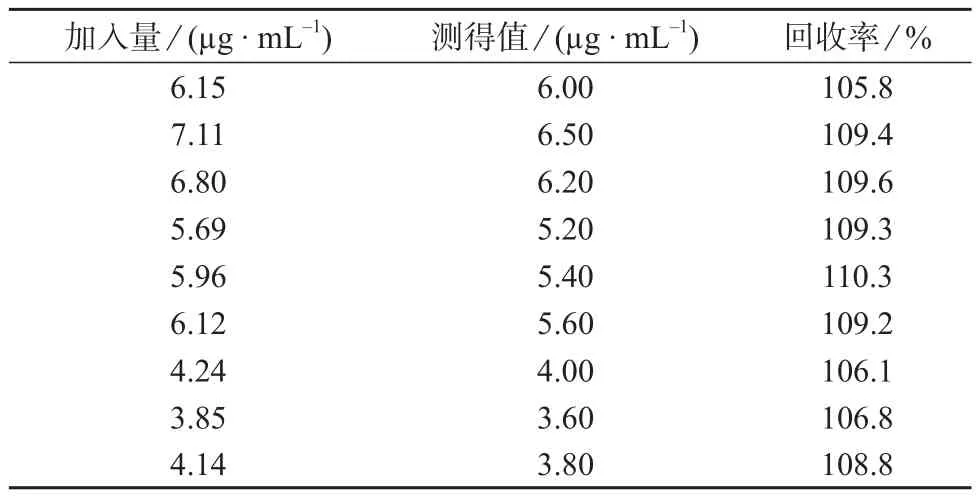
表2 加标回收试验结果
3 结语
采用UPLC BEH C8色谱柱(2.1 mm×50 mm,1.7 μm),建立了超高效液相色谱法测定虾青素的方法。在选定的色谱条件下,虾青素与其它杂质得到良好分离,且该方法流动相的流量小,样品的保留时间短,节约溶剂和时间。该方法具有良好的精密度和准确度,可用于虾青素样品及其有关杂质的高通量检测。
[1] Liu X B,Osawa T. Cis astaxanthin and especially 9-cis astaxanthin exhibits a higher antioxidant activity in vitro compared to the all-trans isomer [J]. Biochemical and Biophysical Research Communications,2007,357: 187–193.
[2] 肖素荣,李京东.虾青素的特性及应用前景[J].中国食物与营养,2011,17(5): 33–35.
[3] 张晓丽,刘建国.虾青素的抗氧化性及其在营养和医药应用方面的研究[J].食品科学,2006,27 (1): 258 –262.
[4] 陶姝颖,明建.虾青素的功能特性及其在功能食品中的应用研究进展[J].食品工业,2012,33(8): 110–115.
[5] Tolasa S,Cakli S,Ostermeter U. Determination of astaxanthin and canthaxanthin in salmonid [J]. European Food Research and Technology,2005(6): 787–791.
[6] 许培雅,郑裕国,沈寅初.分光光度法测定红发夫酵母中虾青素含量]J].浙江工业大学学报,2001,29(2): 120–135.
[7] 姜淼,杨贤庆,李来好,等.高效液相色谱法测定虾壳中的虾青素[J].食品科学,2010,31(20): 371–375.
[8] Mitrowska K,Vincent U,Holst C V. Separation and quantification of 15 carotenoids by reversed phase high performance liquid chromatography coupled to diode array detection with isosbestic wavelength approach [J]. Journal of Chromatography A,2012,1 233: 44–53.
[9] 陈伟珠,张怡评,晋文慧,等.高效液相色谱–光电二极管阵列法测定虾青素的含量[J].化学分析计量,2014,23(1): 24–26.
[10] Miao F P,Lu D Y ,Li Y G,et al. Characterization of astaxanthin esters in Haematococcus pluvialis by liquid chromatography–atmospheric pressure chemical ionization mass spectrometry[J]. Analytical Biochemistry,2006,352: 176–181.
[11] Lacker T,Strohschein S,Albert K. Separation and identification of various carotenoids by C30 reversed-phase high-performance liquid chromatography coupled to UV and atmospheric pressure chemical ionization mass spectrometric detection [J]. Journal of Chromatography A,1999,854: 37–44.
[12] Li B,Zhao H Y,Liu J,et al. Application of ultra-high performance supercritical fluid chromatography for the determination of carotenoids in dietary supplements[J]. Journal of Chromatography A,2015,1 425: 287–292.
[13] Bijttebier S D,Hondt E,Noten B,et al. Ultra high performance liquid chromatography versus high performance liquid chromatography: Stationary phase selectivity for generic carotenoid screening[J]. Journal of Chromatography A,2014,1 332: 46–56.
Determination of Astaxanthin by Ultra Performance Liquid Chromatography
Chen Weizhu, Zhang Yiping, Jin Wenhui, Fang Hua, Hong Zhuan
(Third Institute of Oceanography, State Oceanic Administration, People Republic of China; Engineering Research Center of Marine Biological Resource Comprehensive Utilization, State Oceanic Administration; Fujian Collaborative Innovation Center for Exploitation and Utilization of Marine Biological Resources, Xiamen 361005, China)
A ultra performance liquid chromatography(UPLC) method for determination of astaxanthin was developed. The determination of astaxanthin was performed on a UPLC BEH C8column (50 mm×2.1 mm,1.7 μm). The influence of mobile phase,flow rate and column temperature on the separation of astaxanthin was comprehensively studied. The optimal separation condition was as follows: the mobile phase was methanol–water (volume ratio was 75∶25) with a isocratic elution profile and flow rate of 0.5 mL/min,the detection wavelength was 475 nm, the column temperature was 40℃. The mass concertration of astaxanthin has good linear ralationship with the chromatographic peak area in the range of 0.2–10.0 µg /mL, the correlation coefficient r was 0.998 8. The detection limit was 0.1 µg/mL (S/N=3). The quantitation detection limit was 0.2 µg/mL(S/N=10). The relative standard deviation of determination results was 0.41%(n=6), and the recovery was 105.8%–110.3%. The method is rapid,simple,reliable,and sensitive with a good reproducibility, it can be used for the determination of astaxanthin.
astaxanthin; ultra–performance liquid chromatography (UPLC); rapid determination; separation of impurity
O657.7
A
1008–6145(2016)06–0026–04
10.3969/j.issn.1008–6145.2016.06.006
*福建省科技计划项目(2015N0009);国家重点研发计划项目(2016YFF0201100,2016YFF0201104);海洋生物技术产业化中试技术研发公共服务平台(12PZP001SF10);广东海洋经济发展区域示范项目(GD2012–D01–001)
联系人:陈伟珠;E-mail: wzchen@tio.org.cn
2016–08–12
猜你喜欢
当代水产(2022年4期)2022-06-05
中国饲料(2021年17期)2021-11-02
环境保护与循环经济(2021年7期)2021-11-02
食品安全导刊(2021年20期)2021-08-30
食品安全导刊(2021年20期)2021-08-30
当代水产(2021年6期)2021-08-13
粮食储藏(2021年6期)2021-03-29
落叶果树(2021年6期)2021-02-12
广州化工(2020年20期)2020-11-02
广东饲料(2016年4期)2016-12-01
