
遗传性甲状腺髓样癌MEN 2A家系的临床诊治分析
2016-11-22 12:14张刚郝帅江军文强黄灶明张军罗东林徐琰
中国医药导报 2016年15期
张刚 郝帅 江军, 文强 黄灶明 张军 罗东林 徐琰
1.第三军医大学大坪医院&"战外科研究所乳腺甲状腺血管外科,重庆400042;
2.第三军医大学大坪医院&"战外科研究所泌尿外科,重庆400042
遗传性甲状腺髓样癌MEN 2A家系的临床诊治分析
张刚1郝帅1江军2, 文强2黄灶明2张军2罗东林1徐琰1
1.第三军医大学大坪医院&"战外科研究所乳腺甲状腺血管外科,重庆400042;
2.第三军医大学大坪医院&"战外科研究所泌尿外科,重庆400042
目的探讨遗传性甲状腺髓样癌MEN2A家系的临床诊治特咖。方法回顾性分析1个遗传性甲状腺髓样癌MEN2A家系的2例患者的临床资料,同时进行家系成员RET基因调查。结果第1例患者因“甲状腺髓样癌术后复发”入第三军医大学大坪医院(以下简称“我;”)行右侧甲状腺残《切除术+双侧颈部Ⅵ区淋巴结清扫术,术后第83天复查降钙素恢复正常。第2例患者在我;首次就诊后行甲状腺全切除术+双侧颈部Ⅵ区淋巴结清扫术,术后第47天复查降钙素恢复正常。2例患者术后均未出现声音嘶哑,甲状腺旁腺素随访2个月内均恢复正常。基因检测该家系为RET原癌基因第10外显子第611位咖TGC-TAC杂合错义突变。系谱中发现第2代成员均患病,第3代成员中有50%的成员为携带RET突变基因的未发病者。结论根据临床表现可以确诊MEN2A,结合RET原癌基因突变筛查和进一步系谱调查可以早期诊断出无病RET突变基因携带者;通过家系调查,发现p.Cys611Tyr相关MEN2A家系属于国内首例,该疾病以甲状腺髓样癌和嗜铬细胞瘤发病为特征,不伴有甲状旁腺功能亢进的表现。
遗传性甲状腺髓样癌;MEN 2A;RET原癌基因
甲状腺髓样癌在甲状腺恶性肿瘤中发病率占5%~10%[1],而遗传性甲状腺髓样癌(hereditarymedullary thyroid carcinoma)仅占甲状腺髓样癌中的20%~30%[2-3]。遗传性甲状腺髓样癌MEN2A(multipleendocrine neoplasia type 2A)发病率大<为1/25 000[4]。MEN2A患者中90%出现甲状腺髓样癌,40%~50%的患者可同时出现嗜铬细胞瘤,20%~30%的患者可检查出甲状旁腺功能亢进[4]。临床诊断上MEN2A患者必须要同时合并甲状腺髓样癌和嗜铬细胞瘤[5]。RET原癌基因的错义突变是MEN2A发病的分子基础[6-7]。在肿瘤直径超过1 cm的甲状腺髓样癌患者中有超过70%的患者已经发生颈部淋巴结转移[7];该期患者在生化治愈率上要减少30%以上[8]。因此,早期诊断和手术治疗是治疗甲状腺髓样癌的关键。
查阅文献发现国内未有RET原癌基因p.Cys611Tyr相关MEN2A家系的报道资料,国外对611位咖相关突变的MEN2A家系报道少见,故本研究总结2例家族性甲状腺髓样癌MEN2A患者的临床资料,同时进行系谱调查,进一步探讨RET原癌基因p.Cys611Tyr突变遗传性甲状腺髓样癌MEN2A家系的临床诊治要咖,详细情况汇总如下:
1 象与方法
1.1 研究对象
选取2015年3~8月第三军医大学大坪医院(以下简称“我;”)收治并随访调查的1个家族HMTCMEN2A家系,见图1,调查家族3代共8名成员,其中男性成员4人,女性成员4人。
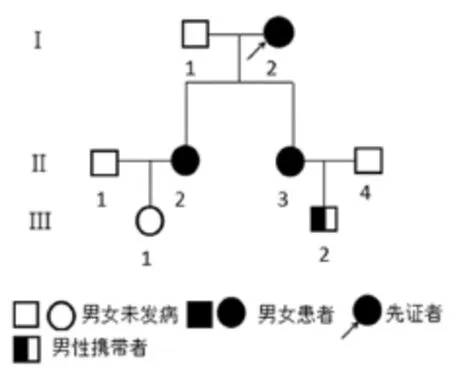
图1 家族性甲状腺髓样癌MEN2A患者系谱
1.2 RET基因检测方法
采用目标区域捕获结合二代高通量测序技术进行分析。
1.3 病理学检查
扪及肿瘤后沿最大径剖开取材,厚度<0.3 cm。4%中性甲醛常规固定、常规脱水后石蜡包埋;4μm厚切片,HE染色后切片观察。
1.4 影像学检查和生化检测
2 果
2.1 临床资料和家系调查结果
2.1.1 第1代成员情况先证者Ⅰ2,老年女性,曾行“双侧嗜铬细胞瘤切除术”,患者既往曾行甲状腺彩超检查提示:双侧甲状腺肿瘤,患者拒绝进一步诊治,行RET原癌基因检测提示:p.Cys611Tyr杂合子突变。Ⅰ1,老年男性,未诉有甲状腺及其他内分泌肿瘤病史,因不在本地未行甲状腺彩超和降钙素检测,未行RET原癌基因检测。
2.1.2 第2代成员情况Ⅱ1和Ⅱ4未诉有甲状腺及其他内分泌肿瘤病史,一直在外务工未回。Ⅱ2,女,43岁,2013年4月因“发现颈前部包块1年余”在当地医院行“甲状腺右侧腺《大部分切除术+左侧甲状腺《切除术”,术后病理提示:双侧甲状腺髓样癌。2014年5月因复发行双侧颈部中央区淋巴结清扫术,术后病理提示:左侧颈部中央区淋巴结(0/5)未见癌组织转移,右侧颈部中央区淋巴结(2/5)查见癌组织转移。我;以“甲状腺髓样癌术后再次复发”收入;。患者2006年因“头痛、头晕”在当地医院检查发现双侧肾上腺病变行“双侧肾上腺嗜铬细胞瘤切除术”。完善检查后遂行右侧甲状腺残《切除术+双侧颈部中央区淋巴结清扫术。Ⅱ3,女,42岁,因“头晕头痛”于2015年3月入;。门诊以“双侧肾上腺嗜铬细胞瘤”收入我;治疗。临床诊断:①双侧肾上腺嗜铬细胞瘤;②左侧甲状腺肿瘤。第1次手术行经腹左侧肾上腺嗜铬细胞瘤切除术,第2次行后腹腔镜下右侧肾上腺嗜铬细胞切除术+甲状腺全切除术+双侧颈部中央区淋巴结清扫术。
2.1.3 第3代成员Ⅲ1,女,19岁,无甲状腺肿瘤及其他内分泌肿瘤病史,基因检测结果:无突变;Ⅲ2,男,18岁,无甲状腺肿瘤及其他内分泌肿瘤病史,甲状腺彩超未见明确病变,患者拒绝抽血行降钙素检测,基因检测报告提示:p.Cys611Tyr杂合子突变。
2.2 Ⅱ2患者辅助检查结果及病理结果
入;后查颈部B超提示:双侧颈部血管旁可见淋巴结,左侧明显(图2a),加彩后可见周围少许血流信号(图2b)。查颈部CT提示:左侧颈部大血管旁可见串珠样淋巴结(图2c)。PET-CT检查无远处转移(图2d)。第245天随访查降钙素提示:160.60 pg/mL;进一步行-DMSA(V)(图2e)提示:左锁骨下血管下方可见一团状增强影,总体表现出延迟减低,结合临床不除外甲状腺髓样癌复发可能。术后病理提示:右侧甲状腺残《未见癌组织,右侧颈部中央区淋巴为纤维结缔组织,左侧颈部中央区淋巴结(3/3)查见癌组织转移(图2f)。
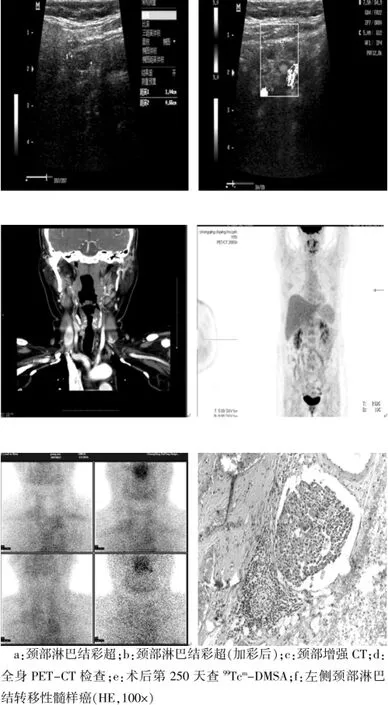
图2 Ⅱ2患者辅助检查结果及病理结果
2.3 Ⅱ3患者辅助检查结果及病理结果
入;后查甲状腺彩超提示:左侧甲状腺中上极可见2.0 cm×1.5 cm的低回声影,内部回声不均匀,内可见粗大的强回声斑(图3a)。CT提示:左侧甲状腺可见2.0 cm×1.5 cm不均匀强化的低密度区,可见粗大的不规则钙化(图3b)。双侧肾上腺可见软组织块状影,边界清楚,密度欠均匀,其大小为右侧4.6 cm×5.5 cm,左侧8.0 cm×5.5 cm,增强见不均匀明显强化(图3c)。第1次术后病理提示:左侧嗜铬细胞瘤(图3d)。第2次术后病理提示:右侧肾上腺嗜铬细胞瘤(图3e)。左侧甲状腺髓样癌(图3f),右侧甲状腺癌灶仅0.3 cm,左侧颈部中央区淋巴结查见(3/6)癌组织;右侧颈部中央区淋巴结未见(0/6)癌组织。
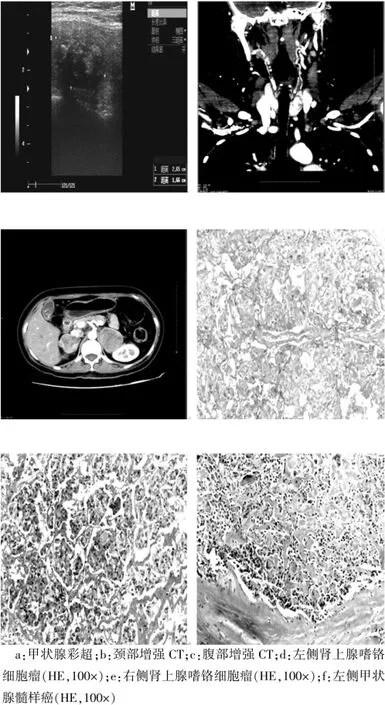
图3 Ⅱ3患者辅助检查结果及病理结果
2.4 家系中第2代成员Ⅱ2、Ⅱ3患者术后随访资料
门诊查甲状旁腺激素和血钙了解甲状旁腺功能恢复情况,查降钙素和癌胚抗原主要了解手术效果及病情变化。Ⅱ2患者术后随访了245 d:甲状旁腺激素和血钙在第42天恢复正常,降钙素水平在第83天随访恢复正常,后期随访发现逐渐升高,具体资料整理见表1。Ⅱ3患者术后随访了138 d:甲状旁腺激素和血钙在第47天恢复正常,降钙素水平在第183天随访恢复正常,Ⅱ3患者自觉降钙素恢复正常,且在外务工,中断随访,遂将现有随访资料整理见表2。

表1 Ⅱ2患者相关血清指标随访情况汇总
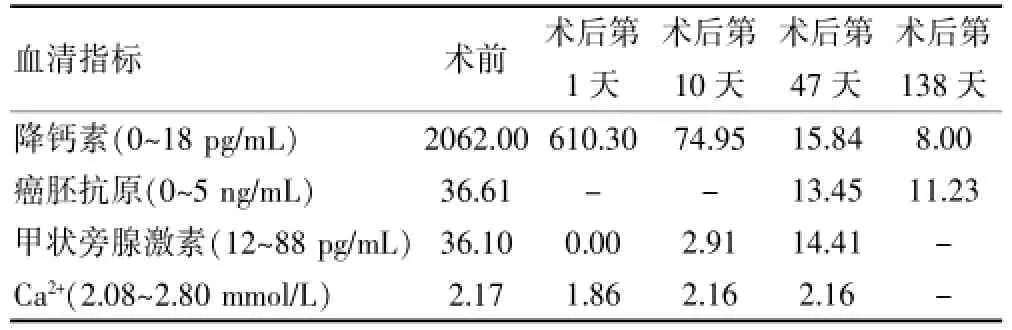
表2 Ⅱ3患者相关血清指标随访情况汇总
2.5 基因检测报告资料汇总
家系中突变患者数为4位,分别为Ⅰ2、Ⅱ2、Ⅱ3、Ⅲ2。突变基因表型均为Cys611Tyr。对应核苷酸序列为c1832。对应核苷酸改变为TGC-TAC。Ⅱ2和Ⅱ3患者RET原癌基因检测的峰图见图4(封三)。
3 论
3.1 家系临床特点与术前辅助检查
Ⅱ2和Ⅱ3患者均有病理报告证实且以甲状腺髓样癌和嗜铬细胞瘤同时发病为特征,符合MEN2A的诊断标准。MEN2A患者典型发病年龄一般在20~40岁[5],女性发病常见[5],该家系中患病成员的临床情况与此大致符合。上述两位患者在各种检查中均未发现甲状旁腺功能亢进的证据,而与甲状旁腺功能亢进密切相关的MEN2A患者RET原癌基因突变常发生在634位咖[9]。临床表现上均以“头昏、头痛”为初诊原因,且均表现为双侧嗜铬细胞瘤发病。该家系中甲状腺髓样癌发病相对嗜铬细胞瘤隐匿,且双侧发病。Ⅱ3患者术前已明确左侧甲状腺髓样癌的诊断,术后病理同时发现隐匿的右侧甲状腺髓样癌(癌灶0.3 cm),此时行RET原癌基因检测虽然能发现基因突变,已错失最佳检测时间。临床实践中,RET基因检查受限于检测技术及其昂贵的检查费用,一般情况下,MEN2A的诊断中,详细询问病史及家族史可以得到有效的线索,通过查降钙素检测、肾上腺皮质激素、甲状旁腺激素、B超和CT检查可以得到初步诊断[10]。
3.2 家系系谱特点分析
家系第1代中Ⅰ2患者发病在家系调查中明确为先证者,RET原癌基因检测发现P.Cys611Tyr突变,且为杂合子突变。该家系第2代成员Ⅱ2与Ⅱ3均患病,第3代中Ⅲ1未发病,检测未发现RET原癌基因突变,Ⅲ2为无病RET原癌基因携带者。家系遗传符合常染色体显性遗传病特咖,系谱特咖为:双亲中有一方Ⅰ2患病,且RET原癌基因检测提示突变为杂合子;第2代成员100%发病,RET原癌基因检测均提示杂合子突变。第3代中无患病成员,有1名成员为无病杂合子突变携带者。家系中女性发病机会比男性概率大,可能与患者家族中未生育多胎有关;总体在家系内表现出连续遗传特咖[11]。临床观察Ⅱ2与Ⅱ3相比表型存在明显差异,也说明杂合子遗传过程中可能受到未知的内外环境影响[11]。
3.3 RET原癌基因突变位点分析
本文涉及的家族性甲状腺髓样癌MEN2A患者均为RET基因p.Cys611Tyr突变。该突变为c.1832 G>A(p.Cys611Tyr),该突变会导致RET基因编码蛋白第611位的半胱氨酸突变为酪氨酸。RET原癌基因p.Cys611Tyr突变相关的家族性甲状腺髓样癌MEN2A家系报道很少,611位咖突变的家族性甲状腺髓样癌合并先天性巨结肠的家系最早在2002年由日本科学家Mikiko Nishikawa报道,家系成员临床表现仅患有甲状腺髓样癌和先天性巨结肠病,分型描述介于MEN2A和FMTC之间,该报道提示的突变位咖为p.Cys611Ser[12],此后国外仅有少量报道,611位咖突变与FMTC和MEN2A两种类型相关[13],进一步单基因分析该位咖突变符合常染色体显性遗传[13]。611位咖突变也与MEN2B类型相关[14],且明确该突变为有害突变[13-15]。国内尚无此位咖相关家系资料报道,因此笔者报道的p.Cys611Tyr突变家系在国内属于首例。
3.4 家系携带者的处理要点
MEN2A患者一旦确诊,早期接受规范化治疗,可以提高临床治愈率,无病RET原癌基因携带者行预防性手术(甲状腺全切术+颈部Ⅵ区淋巴结清扫和甲状旁腺切除同期行自体甲状旁腺移植),可以获得很好的疗效,长期随访可以达到防治遗传性甲状腺髓样癌的目的[16-17]。患者(Ⅲ2)通过筛查发现RET基因611位咖突变。患者已有18岁,但患者及家属拒绝进一步检查和处理。结合诊治体会以及现阶段我国国情,建议无患病RET原癌基因突变携带患者,每年查血清降钙素水平以确定基础降钙素水平,同时结合颈部B超进行临床随访,在患者配合的情况下可择期开展预防性甲状腺切除术。根据美国ATA指南RET突变等级提示,611位咖突变属于B级突变,风险等级较高,在无任何发病的前提下可在5岁后行预防性甲状腺全切除术[7]。根据患者血清降钙素水平及术前颈部B超检查结果综合评估,尽早行甲状腺全切除术,酌情加做Ⅵ区淋巴结清扫术,是Ⅲ2可以获取的最佳治疗方案。
3.5 家系患病成员的手术治疗经验
手术治疗是MEN2A患者的主要治疗手段,MEN2A患者发病情况中甲状腺髓样癌的恶性程度大于嗜铬细胞瘤,在手术顺序上却需先行嗜铬细胞瘤切除术,以避免术后出现肾上腺皮质危象[18]。Ⅱ2患者在外;首诊后意外发现甲状腺髓样癌,可能因技术条件有限以及未行术中冰冻病理检查等原因,导致手术范围不够,并增加了后续手术治疗的复杂性及难度。两例患者术前检查评估未发现颈部侧方淋巴结转移,故未行颈侧方淋巴结清扫术。我;较早应用纳米炭示踪在甲状腺癌术中,取得较丰富的经验,术中应用纳米炭,可使甲状腺及淋巴结黑染,而同侧甲状旁腺和喉返神经未黑染;特别对于经历多次手术的Ⅱ2患者,由于术"粘连严重,解剖层次不清,应用纳米炭示踪,可以仔细辨认、谨慎清除黑染淋巴组织,既可以保证清扫的彻底性,还可防止甲状旁腺及喉返神经损伤[19]。术后2例患者均未出现声嘶等喉返神经损伤表现,甲状旁腺素在随访2个月后均恢复正常,生化指标无异常。说明纳米炭应用有助于术中对于甲状旁腺和喉返神经的保护。
3.6 术后随访
甲状腺髓样癌患者在术后对常规放化疗方案治疗不敏感,术后常规可长期口服左甲状腺素钠维持甲功正常即可[7]。甲状腺髓样癌患者术后常规随访血清降钙素,以测定基线降钙素水平,术后每半年到一年复查1次,正常后每年复查1次,以明确血清降钙素倍增时间,了解病情进展、复发情况[7]。在术后转移灶监测中,降钙素水平的持续升高要比颈部CT和颈部B超发现阳性结果更早、更敏感[20]。第245天随访发现降钙素明显升高,行颈部B超未见明显异常,查-DMSA提示:左锁骨下血管下增强影,结合延迟显像表现及病史,不除外甲状腺髓样癌复发。一般来说髓样癌复发转移,多发生在患侧和对侧颈部淋巴结区域[20],故目前暂予门诊随访。
综上所述,笔者通过RET检测和系谱调查发现了p.Cys611Tyr相关MEN2A家系,属国内首例,该疾病以甲状腺髓样癌和嗜铬细胞瘤发病为特征,不伴有甲状旁腺功能亢进的表现。术后患者随访主要以血清降钙素和颈部B超检查为主,重咖监测患者术后复发转移情况。
[1]CampbellMJ,Seib CD,Gosnell J.Vandetanib and themanagement of advanced medullary thyroid cancer[J].Curr Opin Oncol,2013,25(1):39-43.
[2]Roman S,Lin R,Sosa JA.Prognosis of medullary thyroid carcinoma[J].Cancer,2006,107(9):2134-2142.
[3]Thakker RV.Multiple endocrine neoplasia type 1(MEN1)and type 4(MEN4)[J].Mol Cell Endocrinol,2014,386(1):2-15.
[4]Tang KL,Lin Y,Li LM.Diagnosis and surgical treatment ofmultiple endocrine neoplasia type 2A[J].World JSurg Oncol,2014,12:8.
[5]王军轶.遗传性甲状腺髓样癌家系RET基因种系突变检测分析与临床应用[D].北京:北京协和医学院,2012.
[6]Hansford JR,Mulligan LM.Multiple endocrine neoplasia type 2 and RET:from neoplasia to neurogenesis[J].JMed Genet,2000,37(11):817-827.
[7]Kloos RT,Eng C,Evans D B,et al.Medullary thyroid cancer:management guidelines of the American Thyroid Association[J].Thyroid,2009,19(6):565-612.
[8]Tavares MR,Toledo SP,Montenegro FL,et al.Surgical approach to medullary thyroid carcinoma associated with multiple endocrine neoplasia type 2[J].Clinics,2012,67 Suppl 1:149-154.
[9]Moley JF,Skinner M,GillandersWE,et al.Management of the parathyroid glands during preventive thyroidectomy in patients with multiple endocrine neoplasia type 2[J].Ann Surg,2015,262(4):641-646.
[10]LemosMC,Thakker RV.Multiple endocrineneoplasia type 1(MEN1):analysisof 1336mutations reported in the first decade following identification of the gene[J].Human Mutation,2008,29(1):22-32.
[11]魏雯,刘小莺,姜晓华,等.一多发性内分泌腺瘤病2A型家系的临床调查及基因分析[J].基础医学与临床,2010,30(8):873-876.
[12]Nishikawa M,Murakumo Y,Imai T,et al.Cys611Sermutation in RET proto-oncogene in a kindred withmedullary thyroid carcinoma and Hirschsprung's disease[J].Eur J Hum Genet,2003,11(5):364-368.[13]Machens A,Niccoli-Sire P,Hoegel J,etal.Earlymalignant progression of hereditary medullary thyroid cancer[J].N Engl JMed,2003,349(16):1517-1525.
[14]Hedayati M,Zarif Yeganeh M,Sheikhol Eslami S,et al. Predominant RET germline mutations in exons 10,11,and 16 in Iranian patients with hereditarymedullary thyroid carcinoma[J].JThyroid Res,2011,2011:264248.
[15]Prazeres HJ,Rodrigues F,Figueiredo P,et al.Occurrence of the Cys611Tyr mutation and a novel Arg886Trp substitution in the RET proto-oncogene in multiple endocrine neoplasia type 2 families and sporadic medullary thyroid carcinoma cases originating from the central region of Portugal[J].Clin Endocrinol,2006,64(6):659-666.
[16]Brandi ML,Gagel RF,Angeli A,et al.Consensus:guidelines for diagnosis and therapy of MEN type 1 and type 2[J].JClin EndocrinolMetab,2001,86(12):5658-5671.
[17]Eng C.Mendelian genetics of rare—and not so rare—cancers[J].Annals of the New York Academy of Sciences,2010,1214(1):70-82.
[18]Zhou Y,Zhao Y,Cui B,et al.RET proto-oncogenemutations are restricted to codons 634 and 918 in mainland Chinese families with MEN2A and MEN2B[J].Clin Endocrinol,2007,67(4):570-576.
[19]Tian W,Jiang Y,Gao B,et al.Application of nano-carbon in lymph node dissection for thyroid cancer and protection of parathyroid glands[J].Med Sci Monit,2014,20(24):1925-1930.
[20]Jin LX,Moley JF.Surgery for lymph node metastases of medullary thyroid carcinoma:a review[J].Cancer,2016,122(3):358-366.
Analysis of clinical diagnosis and treatment of a MEN2A pedigree w ith hereditary medullary thyroid carcinoma
ZHANG Gang1HAO Shuai1JIANG Jun2YUAN Wenqiang2HUANG Zaoming2ZHANG Jun2LUO Donglin1XU Yan1
1.Department of Breast,Thyroid,and Vascular Surgery,Research Institute of Surgery,Daping Hospital,Third Military Medical University,Chongqing 400042,China;2.Department of Urology Surgery,Research Institute of Surgery,Daping Hospital,Third Military Medical University,Chongqing 400042,China
Objective To investigate the clinical characteristics of MEN2A patients with hereditary medullary thyroid carcinoma.M ethods The clinical data of 2members of a MEN2A pedigree with hereditarymedullary thyroid carcinoma was analyzed retrospectively,and RET genetic screening in all familymemberswere performed.Results One patient received the residual right thyroid tissue and bilateral cervicalⅥlymph node dissection in Daping Hospital,Third Military Medical University("our hospital"for short)because ofmedullary thyroid cancer recurrence,the calcitonin level returned to normal at 83 days after surgery.Another patient received total thyroidectomy and bilateral cervicalⅥlymph node dissection after the initial diagnosis was confirmed in our hospital,the calcitonin level returned to normal at47 days after surgery.Neither of them appeared hoarseness or loss of voice and both parathyroid hormone returned to normal within 2 months after operation.RET genetic screening showed that heterozygousmissensemutations of TGC to TAC at codon 611 on exon 10 in this pedigree.All two members of the second generation had developed medullary thyroid carcinoma,50%member of the third generation was a asymptomatic RET carrier.Conclusion MEN2A can be diagnosed according to clinicalmanifestations,asymptomatic RET carrier can be diagnosed early through RET genetic screening and further pedigree investigation.Through the investigation of the family,it is found that the p.Cys611Tyr related MEN2A family is the first one in China.The disease is characterized by medullary thyroid carcinoma and pheochromocytoma,not accompanied with themanifestations of hyperparathyroidism.
Hereditary medullary thyroid carcinoma;Multipleendocrineneoplasia type2A;RETproto-oncogene
R736.1
A
1673-7210(2016)05(c)-0030-06
2016-02-13本文编辑:张瑜杰)
张刚(1988-),男,第三军医大学2014级普外科在读硕士研究生;研究方向:甲状腺髓样癌的临床诊治。
徐琰(1976-),男,博士,副主任医师,副教授,硕士生导师;研究方向:甲状腺癌的发病机制。罗东林(1967-),男,博士,主任医师,教授,博士生导师;研究方向:甲状腺癌的发病机制。
猜你喜欢
中国现代医生(2021年9期)2021-07-07
天津医科大学学报(2021年2期)2021-03-29
临床肝胆病杂志(2020年9期)2020-09-28
中国临床医学影像杂志(2019年5期)2019-08-27
现代泌尿生殖肿瘤杂志(2018年5期)2018-11-29
现代仪器与医疗(2017年1期)2017-03-29
西南国防医药(2016年6期)2016-12-01
分子影像学杂志(2015年3期)2015-12-04
癌变·畸变·突变(2014年1期)2014-03-01
河北医科大学学报(2011年3期)2011-03-25
