
单链聚乙烯在石墨烯二维表面结晶的分子模拟
2016-11-21 02:53李延頔何雪莲宋敏驹刘柏平
华东理工大学学报(自然科学版) 2016年5期
李延頔, 何雪莲, 黄 宇, 宋敏驹, 高 睿, 刘柏平
(华东理工大学化工学院,上海市多相结构材料化学工程重点实验室,上海 200237)
单链聚乙烯在石墨烯二维表面结晶的分子模拟
李延頔, 何雪莲, 黄 宇, 宋敏驹, 高 睿, 刘柏平
(华东理工大学化工学院,上海市多相结构材料化学工程重点实验室,上海 200237)
目前对于石墨或石墨烯等片层结构对共聚聚乙烯结晶行为的影响研究相对较少,尚未系统揭示其结晶机制。采用分子动力学模拟方法,研究单链聚乙烯在二维石墨烯表面受限状态下的结晶过程。结果发现,与聚乙烯单链结晶不同,聚乙烯沿石墨烯(001)表面发生表面吸附-链折叠-取向有序形成层状晶体,结晶过程由表面吸附和链折叠共同控制,随等温结晶温度变化分为3个区域。另外,支链不利于结晶,但当支链长度大于10时,即开始发生吸附或折叠进入晶区,与主链形成共晶,从而减缓了结晶度随支链长度而下降的趋势,并且支链主要进入石墨层界面附近的晶区。
石墨烯; 聚乙烯; 分子动力学; 支链; 表面
聚烯烃是世界上应用最广泛的合成树脂材料,其产品广泛应用于工业、农业、运输等国民经济的各个领域[1-2];自从石墨烯(GRA)二维片层材料被发现以来,凭借其特殊的力学、电学和热力学等性能而受到了极大关注,这两者结合具有巨大的工业应用前景。迄今为止,研究人员已对聚烯烃/石墨烯(PE/GRA)复合材料的界面结构、结晶行为及服役性能等进行了大量的实验研究[3-11],但仅采用实验手段往往无法深入观察复合材料的微观界面结构,也不能从分子水平揭示高分子结晶行为与最终性能的关系。如今,分子动力学模拟方法日益广泛地被应用于高分子微观结构的认知研究中,从本质上解析高分子链结构及结晶的微观机理,成为研究高分子结构与性能关系的主要手段之一。
前人已经采用分子动力学方法对聚乙烯(PE)结晶机理进行了大量研究。Kavassalis等[3-4]模拟发现均聚聚乙烯链先发生局部塌缩而后引发链折叠形成片状晶体。最近,Liu等[5]研究了主链长度(以主链中CH2个数计,下同)为10 000的单链聚乙烯结晶,发现主链长度对结晶形态和机理也产生一定的影响。Zhang等[6-7]在模拟中进一步发现当支链长度为10~12时,支链开始进入晶区,与主链形成共晶。
石墨烯二维表面的受限条件会明显影响聚烯烃的结晶行为。Zhao等[8]在实验中发现聚丙烯更倾向于在石墨烯边缘处形成微小片晶,其结晶动力学行为受石墨烯影响很大。Cheng等[9]发现在溶液体系中,聚乙烯首先会在石墨烯表面形成随机分布的棒状晶核,进而生长成约几百纳米大小的片晶,且DSC结果表明随着石墨烯含量增加,聚乙烯的结晶温度及结晶度均增加。Wang等[10]也采用分子动力学模拟方法研究了石墨烯片层引导单链聚乙烯等温结晶行为,发现结晶主要有吸附和取向两步,结晶驱动力主要是石墨烯片层与聚乙烯链间的范德华相互作用。关欣等[11]考察了聚乙烯链在石墨(001)面吸附结晶过程,发现石墨烯表面积大小能够影响吸附层的厚度,但不影响其结晶取向方向。Anastassia等[12]通过模拟发现石墨烯片层附近聚乙烯分子链的密度更大。目前对石墨烯二维表面受限条件下聚乙烯结晶行为的研究尚未更加系统深入地进行,而且大部分聚乙烯产品含有不同长度和含量的支链,而支链对高附加值聚乙烯产品的结晶行为和宏观性能起着决定作用。
本文以均聚和共聚聚乙烯单链模型为研究对象,采用分子动力学模拟方法,研究了石墨烯片层诱导均聚和共聚单链聚乙烯在其表面的吸附结晶行为,考察了支链对结晶行为的影响,并与聚乙烯单链结晶行为进行比较,深入探讨聚乙烯在石墨烯二维表面受限条件下的结晶机理。
1 分子动力学模拟
1.1 模型建立
本文借助分子动力学模拟方法,采用美国Accelrys公司发布的Material studioTM6.1软件建立了均聚和共聚的单链聚乙烯模型(其结构参数如表1所示,其中主链与支链长度均以链中含有的CH2数目计)以及石墨烯片层结构。PE单链采用联合原子模型,主链长度为2 000,相对分子质量为2.8×104,支链均匀分布在主链上。石墨烯片层碳原子数为1 861,并将石墨烯上的碳原子坐标固定,即计算过程中主要考虑PE单链与石墨烯层之间的相互作用。

表1 聚乙烯单链模型结构参数Table 1 Structure parameters of single chain PE models
1.2 模拟方法
采用Material studioTM6.1中的Forcite模块对上述模型进行分子动力学模拟,选用NVT系综(即原子总数N,系统体积V及温度T保持不变,系综需与外界发生能量交换,周期边界足够大),用Andersen方法调节体系温度和压力[13],模拟的基本时间步长为1 fs。本文选用Dreiding II力场[14],力场中总势能(Etotal)可以表示为共价能(如键能Ebond、键角能Eangle、扭转能Etorsion、面相互作用能Eopp等)及非键相互作用(如范德华力Evdw、库仑力Ecoulomb和氢键能EHbond等)之和,即:
Etotal=Ebond+Eangle+Etorsion+Eopp+Evdw+Ecoulomb+EHbond
模拟过程如下:
(1) 首先将优化后的 PE分子链在Dynamics模块中于800 K塌缩成无规线团,并置于距离石墨烯表面小于10-9m的范围内,以使PE分子链和石墨烯发生相互作用。
(2) 模拟均聚PE及均聚PE/GRA复合材料的等温结晶行为:等温结晶时间为3 000 ps,等温结晶温度为300 ~600 K,每隔25 K选取温度点。
(3) 模拟均聚PE及均聚PE/GRA复合材料的非等温结晶行为:降温区间为800~300 K,降温速率分别为0.01,0.1,0.2,0.33,0.5,1,5,10 K/ps。
(4) 模拟共聚PE及共聚PE/GRA复合材料的非等温结晶和等温结晶行为,条件与均聚PE单链相同,并考察支链长度、含量对PE单链及PE/GRA复合材料结晶行为的影响。
结晶度(Xc)计算方法[15]:在模拟高分子结晶过程中经常采用位点序参数(SOP)来衡量体系的结晶程度,其相关计算公式如下:
其中:e表示单位键矢量;i和j是两个相邻原子。SOPk表示对于空间中一个位点k的序参数,将所有位点的SOP值加以平均,即得到整个体系的位点序参数。
同时,根据SOP值可以计算体系的结晶度(Xc),公式如下:
其中:Ncv表示系统中SOP值大于某一数值(本文取SOP峰值约为0.7[15])的位点数,N表示总的位点数。
2 结果与讨论
2.1 概述
前人对聚乙烯结晶机理已有大量的实验和理论研究,但仍主要聚焦于单一聚乙烯体系的非等温和等温结晶行为的研究,以及长、短支链对聚乙烯结晶行为的影响机制。虽然Guan、Anastassia等初步研究了石墨或石墨烯等片层结构对聚乙烯结晶行为的影响,但尚未对其结晶机制进行深入系统的研究,故本文从石墨烯诱导聚乙烯结晶的形态、结晶行为、支链影响等角度系统分析其结晶机理。
2.2 石墨烯诱导PE单链结晶过程的形态变化
通过分子动力学模拟,可以直观地观察到体系随模拟时间的演化过程。图1示出了均聚PE单链的塌缩过程,可以看出,聚乙烯单链先局部塌缩,再整体塌缩形成无规线团。图2和图3分别示出了塌缩后的聚乙烯单链在400 K下的等温结晶过程中的构象变化。可以看到,聚乙烯单链结晶过程是经由自成核-链折叠-有序化过程形成片晶;而在石墨烯二维表面的受限条件下,聚乙烯的结晶形态和过程发生了明显的变化,随时间变化聚乙烯分子链由松散的无规线团结构吸附到石墨烯表面,然后逐渐沿石墨烯(001)表面铺展形成多层有序结构,最后以石墨表面纹理为模板取向形成规则的层状晶体,经历了表面吸附-链折叠-取向有序的结晶过程。
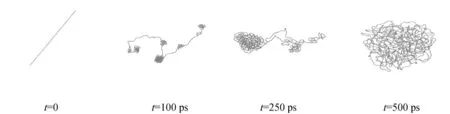
图1 均聚PE单链塌缩过程Fig.1 Collapse process of single chain PE homopolymer

图2 400 K下均聚PE单链等温结晶过程的侧视图Fig.2 Side view of isothermal crystallization process of single chain PE homopolymer at 400 K

图3 400 K下石墨烯诱导均聚PE单链等温结晶过程的侧视及俯视图Fig.3 Side view and top view of isothermal crystallization process of single chain PE homopolymer induced by graphene at 400 K
2.3 石墨烯诱导均聚聚乙烯的结晶行为
2.3.1 等温结晶行为 将塌缩后的PE单链放于石墨烯片层(碳原子数1 861)上进行等温结晶。图4示出了石墨烯诱导PE单链等温结晶的结晶度随温度的变化趋势。可以看到,与文献[16-17]报道的PE单链的等温结晶温度区域相似,石墨烯诱导聚乙烯的等温结晶也可分为I,II和III 3个区域,但机理与PE单链等温结晶相比存在较大差异:当温度高于500 K时(区域I),过冷度较低,分子链运动较快,链沿石墨烯表面纹理铺展并折叠进入晶区的速率较快,但过高的温度阻碍了分子链在石墨烯表面的吸附,此时结晶过程的控制步骤为表面吸附过程,所以随着温度的降低,结晶度迅速增大;当结晶温度在375~500 K时(区域II),分子链运动变慢,石墨烯表面作用变大,PE分子链更易吸附到石墨烯表面,所以结晶由表面吸附和分子链的铺展折叠过程共同决定,随着温度进一步降低,结晶度增大,但增速变缓;当温度降至375 K以下时(区域III),过冷度较大,吸附到石墨烯表面的PE单链运动非常慢,就在吸附位置附近折叠进入晶区,另外,部分PE单链来不及吸附到石墨烯表面,在原地自成核结晶,所以其结晶度迅速降低。
2.3.2 非等温结晶行为 将塌缩后的PE单链模型放置于石墨烯片层(碳原子数1 861)上进行非等温结晶。图5(a)、5(b)示出了不同降温速率下PE单链的非等温结晶构象,图5(c)、5(d)示出了不同降温速率下石墨烯诱导PE单链非等温结晶构象。可以看到,由于石墨烯的诱导作用,相对于PE本体的自成核结晶,石墨烯表面的PE单链可以在更高的温度下发生表面吸附、铺展结晶。图6是均聚PE单链及PE/GRA复合材料非等温结晶的结晶度随降温速率的变化曲线,可以看出,石墨烯的诱导作用对聚乙烯的结晶产生有利影响,在同一降温速率下,其结晶度相对于聚乙烯单链结晶过程明显提高。另外,降温速率加快导致PE/GRA体系的结晶度减小,这是因为随着降温速率增加,分子链来不及在石墨烯表面吸附铺展,只能局部调整使得部分PE链折叠进入晶区,造成片晶结构不规整。

图4 PE/GRA等温结晶的结晶度随结晶温度的变化趋势Fig.4 Xc of isothermal crystallization of PE/GRA at different temperatures

图5 不同降温速率下PE((a)、(b))及PE/GRA((c)、(d))在700 K时的构象Fig.5 Conformation of PE ((a),(b)) and PE/GRA ((c),(d)) with different cooling rates at 700 K
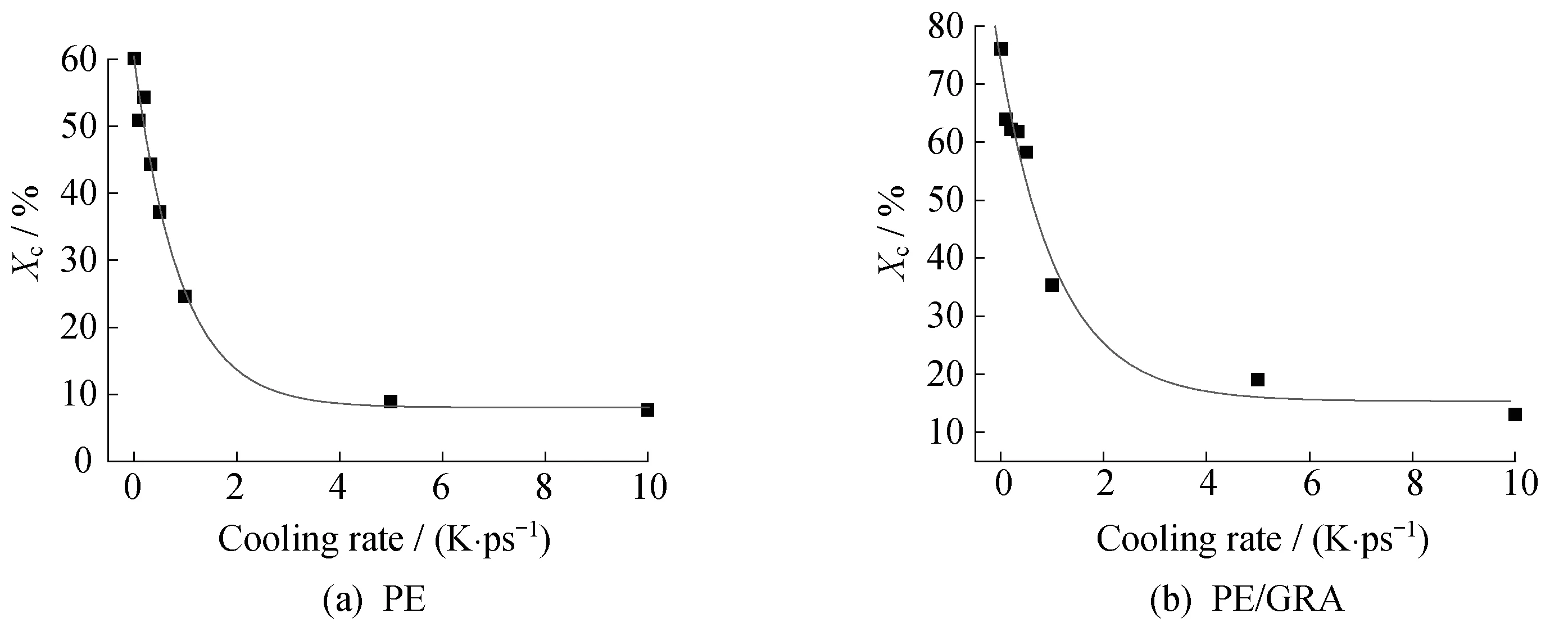
图6 均聚PE及PE/GRA的非等温结晶的结晶度随降温速率的变化趋势Fig.6 Xc of non-isothermal crystallization of PE homopolymer and PE/GRA with different cooling rates
2.4 支链对石墨烯诱导共聚聚乙烯的结晶行为的影响
Zhang等[14-16]从实验和理论的角度研究了短支链对本体聚乙烯结晶机理的影响,本节主要研究了支链长度变化对石墨烯二维片层受限条件下聚乙烯结晶行为的影响机制。
图7示出了当等温结晶温度为400 K,支链数目为10,支链长度分别为0、2、4、6、8、10、12、20、50时,PE单链及石墨烯诱导PE单链等温结晶的结晶度变化。由于在400 K时分子运动速率较快且石墨烯对PE单链有较强表面吸附作用,所以结晶由吸附和铺展折叠两个步骤共同决定。与聚乙烯单链结晶相似,当支链长度为2时,支链作为结晶缺陷被排斥在晶区外,导致形成片晶结构不完整,片晶厚度降低,体系结晶度明显下降;随着支链长度增加,当支链长度为10时,支链开始发生折叠并进入晶区与主链形成共晶,所以体系结晶度降幅减缓。当支链长度继续增加时,PE单链结晶的结晶度仍呈下降趋势,而石墨烯表面的PE单链的结晶度几乎不再降低。

图7 共聚PE及PE/GRA等温结晶的结晶度随支链长度的变化趋势Fig.7 Xc of isothermal crystallization of PE copolymer and PE/GRA with different lengths of chain branch
图8示出了支链长度分别为12和50时共聚PE单链及其在石墨烯表面等温结晶的侧视构象图。可以看到,当支链长度大于10时,在石墨烯二维片层结构的作用下,一方面,更多的支链进入晶区,与主链形成共晶,减缓了结晶度随支链长度下降的趋势;另一方面,约有40%的支链被吸附到石墨烯表面沿其纹理取向结晶。
图9示出了当降温速率为0.1 K/ps,支链数目为10,支链长度分别为0、2、4、6、8、10、12、20、50时,PE单链及石墨烯诱导PE单链非等温结晶的结晶度。可以看到,同等温结晶类似,对于PE单链结晶或石墨烯表面PE单链结晶,当支链长度为10时,部分支链开始折叠进入晶区,与主链形成共晶,体系结晶度降幅减缓。但与等温结晶略有不同,当支链长度继续增加时,本体聚乙烯与石墨烯表面聚乙烯的结晶度均不再降低。
图10示出了支链长度为12和50时共聚PE单链及其在石墨烯表面等温结晶的侧视图。可以看到,当支链长度大于10时,PE及PE/GRA的结晶都进行充分,支链能够充分折叠进入晶区,与主链形成共晶,减缓了结晶度随支链长度下降的趋势。

图8 共聚PE及PE/GRA等温结晶(400 K)的侧视图Fig.8 Side view of isothermal crystallization process of PE copolymer and PE/GRA with chain branch length of 12 and 50 at 400 K

图9 共聚PE及PE/GRA非等温结晶的结晶度随支链长度的变化趋势Fig.9 Xc of non-isothermal crystallization of PE copolymer and PE/GRA with different lengths of branch chain

图10 共聚PE及PE/GRA非等温结晶(0.1 K/ps)的侧视图(支链长度为12和50)Fig.10 Side view of non-isothermal crystallization process of PE copolymer and PE/GRA with branch chain length of 12 and 50 with cooling rate of 0.1 K/ps
另外,支链数目对PE/GRA结晶的影响规律与支链数目对PE单链结晶的影响规律一致,随着支链数目的增加,结晶度均呈下降趋势。
3 结 论
本文采用分子动力学模拟方法研究了PE单链在石墨烯二维片层受限条件下的结晶机理,并考察了结晶温度、降温速率、支链长度及数目等对结晶行为的影响。
(1) 与PE单链结晶不同,在石墨烯二维片层的受限条件下,聚乙烯无规线团逐渐吸附到石墨烯表面,沿石墨烯(001)表面铺展,并以石墨烯表面纹理为模板取向形成规则的层状晶体,经历了表面吸附-链折叠-取向有序的结晶过程
(2) 石墨烯二维片层表面的PE单链等温结晶可分为I,II和III 3个区域,但其机理与PE单链结晶存在较大差异:当温度大于500 K时(区域I),结晶控制步骤为表面吸附过程,随着温度的降低,结晶度迅速增大;当结晶温度为375~500 K时(区域II),结晶由表面吸附和PE链的铺展折叠过程共同控制,随着温度进一步降低,结晶度增大,但增速变缓;当温度降至375 K以下时(区域III),过冷度较大,以PE链铺展折叠为结晶控制步骤,不利于形成规则晶体结构,结晶度迅速降低。
(3) 支链对于聚乙烯单链及其在石墨烯片层上结晶均有一定抑制作用,都是在支链长度为10时,支链开始折叠进入晶区与主链形成共晶,但是机理却不相同,且影响程度略有差异。对于等温结晶过程,在石墨烯二维片层结构受限条件下,一方面更多的支链进入晶区,与主链形成共晶,减缓了结晶度随支链长度下降的趋势;另一方面,约有40%的支链吸附到石墨烯表面沿其纹理取向结晶。另外,聚乙烯单链在石墨烯表面结晶的结晶度随着支链数目的增加而减小。
[1] 陈乐怡.世界聚烯烃工业的发展趋势[J].合成树脂及塑料,2008,25(1):1-7.
[2] 乔金樑.聚烯烃材料的研究开发进展[J].中国材料进展,2012,31(2):33-37.
[3] KAVASSALLIS T A, SUNDARARAJAN P R.A molecular-dynamics study of polyethylene crystallization[J].Macromolecules,1993,26(16):4144-4150.
[4] SUNDARARAJAN P R,KAVASSALIS T A.Molecular dynamics study of polyethylene chain folding:The effects of chain length and the torsional barrier[J].Journal of the Chemical Society,Faraday Transactions,1995,91(16):2541-2549.
[5] GAO Rui,HE Xuelian,ZHANG Haiyang,etal.Molecular dynamics study of the isothermal crystallization mechanism of polyethylene chain:Combined effects of chain length with temperature[J].Journal of Molecular Modeling,2016,DOI:10.1007/s00894-016-2931-2.
[6] ZHANG Xiubin,LI Zesheng,LU Zhongyuan,etal.Roles of branch content and branch length in copolyethylene crystallization:Molecular dynamics simulations[J].Macromolecules,2002,35(1):106-111.
[7] ZHANG Xiubin,LI Zesheng,YANG Hua,etal.Molecular dynamics simulations on crystallization of polyethylene copolymer with precisely controlled branching[J].Macromolecules,2004,37(19):7393-7400.
[8] ZHAO Songmei,CHEN Fenghua,HUANG Yingjuan,etal.Crystallization behaviors in the isotactic polypropylene/graphene composites[J].Polymer,2014,55(16):4125-4135.
[9] CHENG Shan,CHEN Xi,GRACE H,etal.Reduced graphene oxide-induced polyethylene crystallization in solution and nanocomposites[J].Macromolecules,2012,45(2):993-1000.
[10] WANG Lizhi, DUAN Lili.Isothermal crystallization of a single polyethylene chain induced by graphene:A molecular dynamics simulation[J].Computational and Theoretical Chemistry,2012,1002:59-63.
[11] 关欣,杨华,赵小军,等.聚乙烯在石墨表面结晶的分子动力学模拟[J].分子科学学报(中英文版),2009,25(6):390-393.
[12] RISSANOU A N,POWER A J,HARMANDARIS V.Structural and dynamical properties of polyethylene/graphene nanocomposites through molecular dynamics simulations[J].Polymers,2015,7(3):390-417.
[13] ANDERSEN H C.Molecular dynamics simulations at constant pressure and/or temperature[J].The Journal of Chemical Physics,1980,72(4):2384-2393.
[14] MAYO S L,OLAFSON B D,GODDARD W A.Dreidin:A generic force field for molecular simulations[J].Journal of Physical Chemistry,1990,94(26):8897-8909.
[15] YU Xiang,KONG Bin,YANG Xiaozhen.Molecular dynamics study on the crystallization of a cluster of polymer chains depending on the initial entanglement structure[J].Macromolecules,2008,41(18):6733-6740.
[16] HOFFMAN J D, MILLER R L.Kinetic of crystallization from the melt and chain folding in polyethylene fractions revisited:Theory and experiment[J].Polymer,1997,38(13):3151-3212.
[17] ARMISTEAD J P, HOFFMAN J D.Direct evidence of regimes I,II,and III in linear polyethylene fractions as revealed by spherulite growth rates[J].Macromolecules,2002,35(10):3895-3913.
Molecular Simulation of Single Shain Polyethylene Crystallization on Two-Dimensional Surface of Graphene
LI Yan-di, HE Xue-lian, HUANG Yu, SONG Min-ju, GAO Rui, LIU Bo-ping
(Shanghai Key Laboratory of Multiphase Materials Chemical Engineering,School of Chemical Engineering,East China University of Science and Technology,Shanghai 200237,China)
Few studies on the influence of lamellar structure like graphite or graphene on crystallization behavior of PE copolymer were reported,and the crystallization mechanism had not yet revealed systematically.In this paper,the crystallization process of single chain PE induced by graphene surface was investigated with molecular dynamics simulation.It turned out that PE tended to crystalize into lamellar structure on the surface of graphene (001) through surface absorption-chain folding-orientation process.This process,controlled by both of surface adsorption and chain folding,could be divided into three temperature regimes in the progress of isothermal crystallization.In addition,chain branches restrained crystallization,however,they would be adsorbed into the graphene surface or folded into crystalline field to co-crystallize with main chain when the length of chain branch was more than 10.This transformation slowed down the trend that crystallinity decreased with the length of chain branch.Moreover,chain branches tended to fold into crystalline field near the graphene surface.
graphene; polyethylene; molecular dynamics; chain branch; surface
1006-3080(2016)05-0594-07
10.14135/j.cnki.1006-3080.2016.05.002
2016-02-25
国家自然科学基金(51573048)
李延頔(1990-),男,山东人,硕士生,研究方向为聚烯烃高分子材料。
何雪莲,E-mail:hexl@ecust.edu.cn
TQ536.1
A
猜你喜欢
中国海洋大学学报(自然科学版)(2019年2期)2019-12-07
天水师范学院学报(2018年5期)2018-12-04
现代检验医学杂志(2016年4期)2016-11-15
现代检验医学杂志(2016年5期)2016-08-20
江苏农业科学(2015年1期)2015-04-17
科技创新导报(2014年34期)2015-01-13
北京航空航天大学学报(2014年1期)2014-12-19
中国机械工程(2012年15期)2012-07-25
中国麻业科学(2011年5期)2011-12-05
中国机械工程(2010年9期)2010-06-04
