
盐酸鲁拉西酮的合成工艺研究
2016-11-19 07:15:11李玉龙陈年根潘勤鹤张万科
广州化工 2016年20期
李玉龙, 陈年根, 潘勤鹤, 张万科
(1 海南医学院高等职业技术学院,海南 海口 571199;2 海南大学材料与化工学院,海南 海口 5702283;3 海南医学院药学院,海南 海口 571199)
盐酸鲁拉西酮的合成工艺研究
李玉龙1,2, 陈年根3, 潘勤鹤2, 张万科3
(1 海南医学院高等职业技术学院,海南 海口 571199;2 海南大学材料与化工学院,海南 海口 5702283;3 海南医学院药学院,海南 海口 571199)
合成了抗精神分裂药物盐酸鲁拉西酮。以苯并异噻唑-3(2H)-酮为原料,经氯代、与无水哌嗪缩合得3-(哌嗪-1-基)苯并异噻唑,其和(1R, 2R)-环己-1,2-二甲醇二甲磺酸酯反应得反式-3aR, 7aR-八氢异吲哚啉-2-螺-1’-[4’-(1, 2-苯并异噻唑-3-基)哌嗪]甲磺酸盐,再和顺-降冰片烷-外-2,3-二甲酰亚胺反应得鲁拉西酮,最后经成盐得盐酸鲁拉西酮。目标化合物经光谱确证,三步收率为42%(以苯并异噻唑-3(2H)-酮计)。反应条件温和、操作简便,各步原料价格低廉,采用的工艺操作简便易行。
盐酸鲁拉西酮;合成工艺;抗精神分裂药物
盐酸鲁拉西酮(Lurasidone Hydrochloride),化学名为(3aR,4S,7R,7aS)-2-[(1R, 2R)-2-[4-(1,2-苯并噻唑-3-基)哌嗪-1-基甲基]环己烷]六氢-4,7-亚甲基-1H-异吲哚-1,3(2H)-二酮盐酸盐,为白色或类白色结晶粉末。本品在甲醇中略溶,在乙醇中微溶,在丙酮中极微溶解,在水中几乎不溶。盐酸鲁拉西酮是由日本住友制药公司开发的具有双重作用的新型抗精神病药物,它对5-HT2A受体和多巴胺D2受体均具有高度亲和力,对精神病患者的阳性和阴性症状均具有显著疗效。该药于2010年10月28日经美国FDA批准在美国上市[1-3]。
文献[4-6]报道的盐酸鲁拉西酮合成工艺较多,由盐酸鲁拉西酮的结构逆分析可知, 鲁拉西酮盐酸盐结构片段主要有三部分组成:外-二环[2.2.1]-庚烷-2,3-二酰亚胺部分、环己烷衍生物和3-哌嗪-1,2-苯并噻唑部分。在文献的基础上,通过改变其不适应工业化生产的步骤,在打通路线的基础上对其工艺作适当的优化,期望通过实验室的小试研究,为盐酸鲁拉西酮的工业化生产提供理论依据和可行的工艺参数。合成路线见图1。
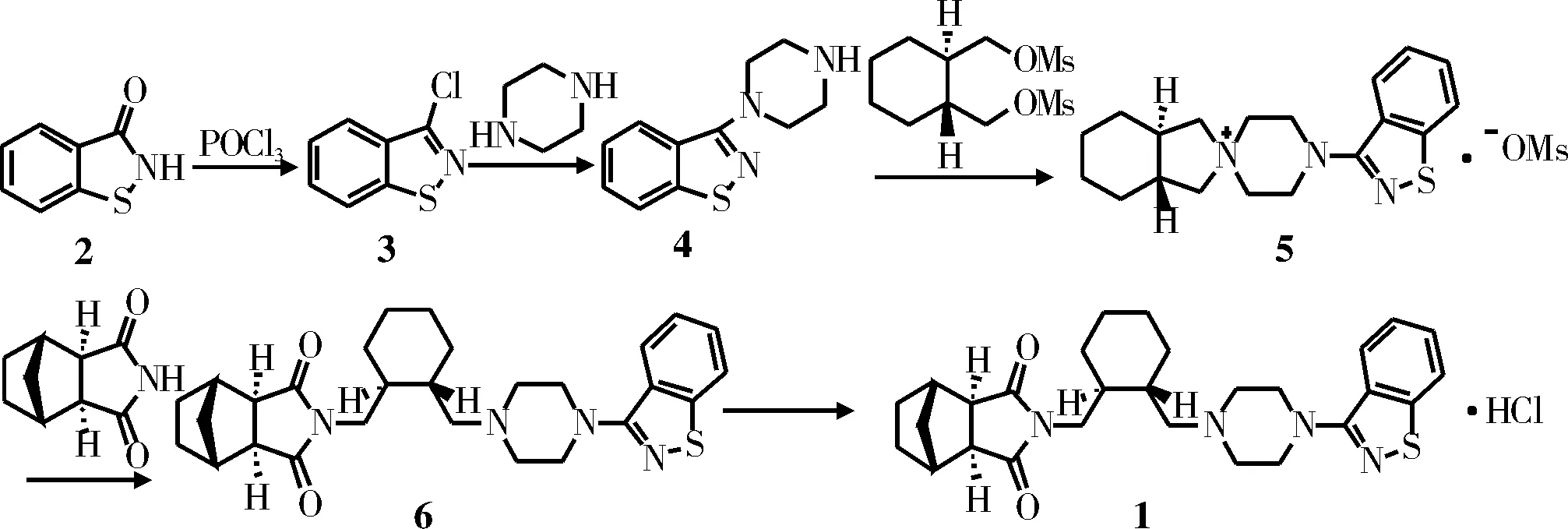
图1 盐酸鲁拉西酮的合成路线
1 实 验
1.1 仪器与试剂
X4显微熔点测定仪;Bruker-AV-400型核磁共振仪;Bruker microTOF-QⅡ高分辨质谱;Thermo Nicolet-5700型傅里叶变换红外光谱仪。
所用试剂为市售工业品或化学纯。
1.2 操作步骤
1.2.1 3-氯苯并异噻唑(3)的合成
1 L三颈瓶中加入苯并异噻唑-3(2H)-酮(2, 75.6 g, 0.5 mol),三氯甲烷(400 mL),搅拌使固体溶解,降温至0~5 ℃,滴加三氯氧磷(168.7 g, 1.1 mol),滴毕,升温至回流反应12 h。冷却至室温,缓慢倾入冰水混合物中,分液,饱和碳酸氢钠洗涤,饱和食盐水洗涤,无水硫酸钠干燥,过滤,减压蒸除溶剂得淡黄色固体78.1 g,收率92.1%。mp: 38~39 ℃(文献[6],mp: 35~36 ℃)。
ESI-MS(m/z):168[M-H]。
1.2.2 3-(哌嗪-1-基)苯并异噻唑(4)的合成
1 L三颈瓶中加入3-氯苯并异噻唑(3, 67.9 g, 0.4 mol),无水哌嗪(86.1 g,1 mol),叔丁醇(300 mL),氮气保护下回流反应24 h。反应体系中加入冰水(500 mL)和50%的氢氧化钠溶液,调节pH大于10,二氯甲烷萃取,饱和食盐水洗涤,无水硫酸钠干燥,减压蒸除溶剂,石油醚重结晶,得黄色固体62.5 g,收率71.3%。mp: 85~87 ℃(文献[6],mp: 83~85 ℃)。
1H-NMR(400 MHz, DMSO-d6) δ: 2.32(br, 1H), 2.87(m, 4H), 3.31(m, 4H),7.39(m, 1H), 7.52(m, 1H), 8.01(m, 2H)。ESI-MS(m/z):220[M+H]。
1.2.3 反式-3aR, 7aR-八氢异吲哚啉-2-螺-1’-[4’-(1,2-苯并异噻唑-3-基)哌嗪]甲磺酸盐(5)的合成
1 L三颈瓶中加入3-(哌嗪-1-基)苯并异噻唑(4, 54.8 g, 0.25 mol)、(1R, 2R)-环己-1,2-二甲醇二甲磺酸酯(78.9 g, 0.26 mol),乙腈(300 mL),无水碳酸钠(61.2 g, 0.58 mol),氮气保护下搅拌回流反应10 h,抽滤,减压蒸除溶剂,加入乙酸乙酯,抽滤,干燥得黄色固体86.1 g,收率81.3%。mp: 225~227 ℃(文献[4],mp: 224~227 ℃)。
1H-NMR(400 MHz, DMSO-d6) δ: 1.21(m, 4H), 1.83(m, 4H), 1.91(m, 2H), 2.30(s, 3H), 3.23(m, 2H), 3.76(m, 8H), 3.96(m, 2H), 7.51(m, 1H), 7.61(m, 1H), 8.13(m, 1H), 8.20 (m, 1H)。
1.2.4 鲁拉西酮(6)的合成
1 L三颈瓶中加入反式-3aR, 7aR-八氢异吲哚啉-2-螺-1′-[4′-(1,2-苯并异噻唑-3-基)哌嗪]甲磺酸盐(5, 63.5 g, 0.15 mol),顺-降冰片烷-外-2,3-二甲酰胺(37.2 g, 0.225 mol),无水碳酸钾(41.4 g, 0.3 mol),N,N-二甲基甲酰胺(500 mL),氮气保护下回流反应3 h,冷却至室温,倒入纯化水中,加入二氯甲烷,分液,有机相纯化水洗涤,饱和食盐水洗涤,无水硫酸钠干燥,抽滤,减压蒸除溶剂,加入乙腈,搅拌析晶,抽滤,干燥得类白色固体61.2 g,收率82.7%。ESI-MS(m/z):493[M+H]。
1.2.5 盐酸鲁拉西酮(1)的合成
500 mL三颈瓶中加入鲁拉西酮50 g,丙酮150 mL,加热溶解,过滤,滴加浓盐酸,逐渐析出大量固体,过滤,干燥,得白色固体47.6 g,收率95.2%。
IR(KBr)cm-1: 3065, 2935, 2914, 2872, 2850, 2310, 2284, 2257, 2224, 1761, 1686, 1562, 1503, 1446, 1429, 1182, 778, 741.1H-NMR(400 MHz,DMSO-d6) δ: 12.51(br, 1H), 7.856~7.825(t, 2H), 7.52~7.49(t, 1H), 7.41~7.38(t, 1H), 4.36-4.18(m, 2H), 4.10~4.05(m, 2H), 3.60-3.45(m, 5H), 3.21~3.19(m, 2H), 2.89(m, 1H), 2.68(s, 2H), 2.65(s, 2H), 2.33-2.31(d, 1H), 1.80-1.78(d, 1H), 1.72-1.66(m, 5H), 1.50-149(d, 1H), 1.35~1.34(m, 3H), 1.30~1.22(m, 3H), 1.11-1.06(m, 2H).13C-NMR(400 MHz,DMSO-d6) δ: 179.49, 161.62, 152.93, 128.00, 127.36, 124.48, 123.35, 120.77, 61.68, 52.89, 49.98, 48.65~48.63, 46.26, 46.17, 41.67, 39.88~39.74, 34.53, 33.36, 31.36, 29.86, 27.98, 24.39, 24.32。ESI-MS(m/z):493[M+H]。
2 结果与讨论
2.1 3-氯苯并异噻唑(3)的合成
本步骤大多采用以三氯氧磷为反应物和溶剂,生成的杂质多,且后处理困难。研究过程中发现,采用在卤代溶剂中反应,后处理相对简单,且生成的杂质较少。
2.2 3-(哌嗪-1-基)苯并异噻唑(4)的合成
文献[6]报道了3-(哌嗪-1-基)苯并异噻唑(4)的生成机理:首先是低温下异噻唑的开环,生成一个氰基,同时哌嗪基接到硫原子处,接着高温下第二分子哌嗪在碳氮三键处进行加成,前两步是快速步骤;最后一步为控制步骤:硫上的哌嗪基与氮上的氢原子结合成哌嗪脱去,然后又形成异噻唑环得到产物。

从反应机理来看,哌嗪在反应中发挥着两个方面的作用,一方面作为反应物,另一方面作为缚酸剂与脱下来的氯离子结合,使得反应能继续进行。考虑到应避免生成哌嗪双取代的产物,哌嗪的量应该过量。文献报道了不用溶剂直接将3-氯-苯并异噻唑与无水哌嗪进行反应的方法,但哌嗪会在实验温度下升华并在冷凝管上端冷凝,使得反应物比例发生改变,影响反应的收率,因而反应必须在使用特殊的反应器中进行。由于反应在无溶剂条件下不易操作,考虑采用溶剂的方法改进反应条件,采用加入溶剂叔丁醇的方法,反应能顺利进行。
2.3 反式-3aR, 7aR-八氢异吲哚啉-2-螺-1′-[4′-(1,2-苯并异噻唑-3-基)哌嗪]甲磺酸盐(5)的合成
本反应的机理是在碱性条件下,(1R, 2R)-环己-1,2-二甲醇二甲磺酸酯和3-(哌嗪-1-基)苯并异噻唑发生亲核取代并季铵化。实验中比较了二者的投料比、碱的选择、溶剂等,发现(1R, 2R)-环己-1,2-二甲醇二甲磺酸酯、3-(哌嗪-1-基)苯并异噻唑和碳酸钠的摩尔比为1.04:1.0:2.23时,反应较好,不同溶剂的反映情况,总体是在乙腈中原料几乎能消耗完全,且产品中杂质较少。
2.4 鲁拉西酮(6)的合成
本步骤的反应类似于盖布瑞尔伯胺合成反应,溶剂和反应体系的碱性对其有重要影响,试验中比较了不同的溶剂和不同的碱,发现N,N-二甲基甲酰胺和碳酸钾体系能够得到较好质量的产品。
2.5 盐酸鲁拉西酮(1)的合成
盐酸鲁拉西酮为难溶性口服固体制剂,晶型对其生物利用度有很大影响。采用丙酮为结晶溶剂,晶型和文献报道的药用晶型一致,且收率高,产品色泽和质量较好。
3 结 论
本文的目标化合物的合成以苯并异噻唑-3(2H)-酮为原料,经氯代、缩合、成季铵盐、开环、成盐酸盐制得。工艺操作简便易行,中间体易纯化,三步收率为42%(以苯并异噻唑-3(2H)-酮计),适宜中试放大及工业化生产。
[1] 汪超(编译), 孙铁民(审校). 盐酸鲁拉西酮(lurasidone hydrochloride)[J]. 中国药物化学杂志,2011, 21(3):248.
[2] 史菁菁,葛渊源,封宇飞,等.2010年美国FDA批准的新药[J].中国新药杂志, 2011, 20(3):197-199.
[3] 杨臻峥编译, 孙大柠审校. 抗精神病药Lurasidone Hydrochloride[J].药学进展, 2009, 33(2):91-193.
[4] Fujio A, Mayumi TS, Ikutaro S, et al.Imide derivatives, and their production and use[P]. EP0314098A2,1987-10-26.
[5] Muto M, Tanno N.Process for producing imide compound [P].WO2005009999 A1,2005-02-03.
[6] Fox DE, Lambert JF.Processes and intermediates for preparing 3-(1-piperazinyl)-1,2-benzisothiazole[P]. EP0874834 B1,2000-12-13.
Study on Process of Lurasidone Hydrochloride
LIYu-long1,2,CHENNian-gen2,PANQin-he2,ZHANGWan-ke2
(1 School of Higher Vocational and Technical Education, Hainan Medical College, Hainan Haikou 571199;2 College of Materials and Chemical Engineering, Hainan University, Hainan Haikou 570228;3 School of Pharmacy, Hainan Medical College, Hainan Haikou 571199, China)
The synthetic process of Lurasidone Hydrochloride, an atypical antipsychotics which had high affinity with 5-HT2A receptor and the dopamine D2receptor, was studied. The key intermediate 3-(piperidin-1-yl)benzisothiazole was synthesized from benzisothiazole-3(2H)-one as raw matercialviachlorinated reaction, condensation reaction with anhydrous piperazine. The intermediate trans-3aR, 7aR-octahydro-isoindol-2-spiro-1’-[4’-(1,2-benzisothiazol-oxadia-zol-3-yl) piperazine]methanesulfonate was synthesized from 3-(piperidin-1-yl)benzisothiazole and (1R, 2R)-cyclohex-1,2-dimethanol dimethyl sulfonic acid ester by condensation reaction.The free base, lurasidone, was obtained from trans-3aR, 7aR-octa-hydroisoindol-2-spiro-1’-[4’-(1,2-benzisothiazol-oxadia-zol-3-yl)piperazine] methanesulfonate and cis-norbornane-exo-2,3-dicarboxylic amide by condensation reaction. The target compound, lurasidone hydrochloride, was obtained by salification. Results whowed that the total yield of three steps was 42%,and the structure of final compoud was confirmed bythe spectrum. The process was simple and easy for industrial production.
lurasidone hydrochloride; synthetic process; atypical antipsychotics
李玉龙(1984-),男,实验师,药学专业,研究方向为药物学。
潘勤鹤(1980-),男,教授,无机化学专业,研究方向为无机合成化学与功能配位化学。
R914.5
A
1001-9677(2016)020-0061-03
猜你喜欢
临床儿科杂志(2023年8期)2023-09-29 09:38:52
证券市场周刊(2023年16期)2023-06-30 12:14:33
科技创新导报(2022年23期)2022-04-04 03:09:54
云南化工(2021年7期)2021-12-21 07:27:22
昆明医科大学学报(2021年10期)2021-12-02 03:24:38
作文周刊·小学四年级版(2018年40期)2018-04-09 08:20:04
合成化学(2015年10期)2016-01-17 08:56:44
郑州大学学报(工学版)(2015年1期)2015-03-24 00:55:36
中国当代医药(2015年30期)2015-03-01 02:08:09
应用化工(2014年7期)2014-08-09 09:20:26
