
有机染料敏化太阳电池中激发态弛豫和电子注入的超快光谱研究
2016-11-18 07:29:15林李阳陈淑张静张
物理化学学报 2016年1期
杨 林李 阳陈 淑张 静张 敏,*王 鹏
(1中国科学院长春应用化学研究所,高分子物理与化学国家重点实验室,长春 130022;2中国科学院大学,北京 100049)
有机染料敏化太阳电池中激发态弛豫和电子注入的超快光谱研究
杨 林1,2李 阳1,2陈 淑1,2张 静1张 敏1,*王 鹏1
(1中国科学院长春应用化学研究所,高分子物理与化学国家重点实验室,长春 130022;2中国科学院大学,北京 100049)
为了实现窄能隙有机光敏剂的理性设计,有必要全面理解发生在二氧化钛/染料/电解质复杂界面的激发态演化动力学。本文通过构建分别以苯并噻二唑-苯甲酸(BTBA)和吡啶并噻二唑-苯甲酸(PTBA)为电子受体的有机给受体染料,借助超快瞬态吸收光谱测量与理论模拟,我们发现在实际的二氧化钛/染料/电解质界面存在激发态多步弛豫与多态电子注入的过程。密度泛函理论及含时密度泛函理论计算表明,二氧化钛表面的光激发产生的“热”激发态染料分子会通过分子片段间的扭转运动发生显著的多步结构弛豫,最终形成共轭骨架具有醌式结构、更加平面化的平衡构型。通过对飞秒瞬态吸收光谱进行目标分析,我们发现相对于以苯并噻二唑-苯甲酸为电子受体的染料,以吡啶并噻二唑-苯甲酸为电子受体的染料呈现出较慢的电子注入速率与较短的激发态寿命,导致总的电子注入产率较低,给出了基于该染料所制备的太阳电池的外量子产率峰值低的原因。
太阳电池;有机染料;界面;激发态;电荷转移
(1State Key Laboratory of Polymer Physics and Chemistry, Changchun Institute of Applied Chemistry, Chinese Academy of Sciences, Changchun 130022, P. R. China;2University of Chinese Academy of Sciences, Beijing 100049, P. R. China)
1 lntroduction
In response to the growing global energy crisis, photoelectric conversion technologies characteristic of ecofriendly and low-cost have attracted widespread attention. In this regards, relentless innovative efforts have been devoted to dye-sensitized solar cells (DSCs)1based on a wide band gap semiconductor in conjunction with a photosensitizer and an electrolyte. A photosensitizer plays a vital role in determining the cell performance by influencing light absorption, carrier generation, and charge recombination2–11. In the past two decades, taking account of the resource scarcity of ruthenium polypyridines and the inferior stability of zinc porphyrins, metal-free organic dyes have attracted a large amount of research passion in virtue of unique properties such as plentiful raw materials, gorgeous colors, and flexible molecular tailoring, affording by far the highest power conversion efficiency of 14.3% in DSCs12.
It is well recognized that narrowing the energy gap of donoracceptor (D-A) dye molecules is one of the basic strategies to augmented light absorption at desirable longer wavelengths. In this regard, the electronic structure of an electron acceptor has profound effects on the light harvesting capacity of a sensitizer by impacting the molecular energy levels, and some interfacial charge transfer dynamic processes such as excited state relaxation and electron injection. This recognition has guided some efforts in designing D-A organic dyes characteristic of various electron acceptors. Recently, benzothiadiazole-benzoic acid(BTBA) has been employed as an electron acceptor to construct metal-free D-A dyes13and zinc porphyrin complexes14, bringing forth crucial progress on power conversion efficiencies.
In this paper, we have first synthesized a new metal-free D-A dye (LY-1, Fig.1) by attaching the BTBA segment to triphenylamine-thienodioxepine unit. To reduce the energy-gap, we have further employed a more electron-withdrawing unit pyridothiadiazole-benzoic acid (PTBA) to replace BTBA and constructed other new dye LY-2 (Fig.1). Our preliminary experiments have shown that there is much lower external quantum efficiency (EQE) summit for LY-2, which has also been observed in many of D-A infrared dyes with small energygaps. The key factor may be the low electron injection yield(ϕei) resulting from the short excited state lifetimes of these organic dyes, which has not received wide recognition. Moreover, the excited state dynamics have only attracted very little attention15–21, resulting in an unclear mechanism of electronic injection dynamics, which cannot provide a rational guide in the further design of narrow energy-gap organic dyes in DSCs. Thereby, based upon these two new dyes, herein we will take a close look at the structure related dynamics of excited state evolution and charge transfer occurring at the titania/dye/electrolyte interface by joint theoretical calculations and experimental measurements.
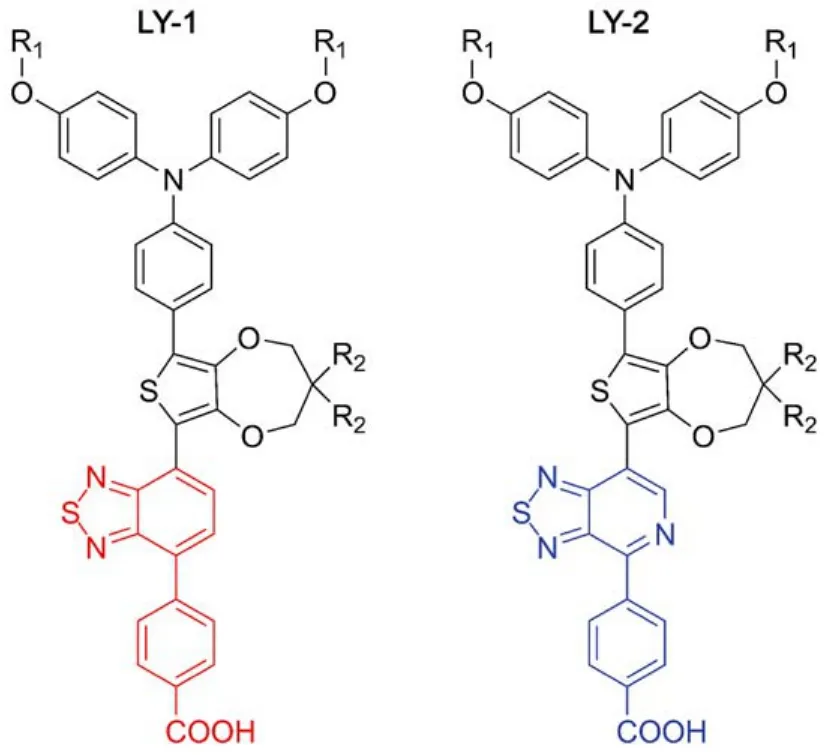
Fig.1 Chemical structures of LY-1 and LY-2, characteristic of BTBA (red) and PTBA (blue)
2 Experimental
2.1 Materials
Chloroform (99.9%), ethanol (99.9%), acetonitrile (99.9%), tetrahydrofuran (THF, 99.9%), lithium bis(trifluoromentylsulfonyl)imide (LiTFSI, 99.9%), 1-ethyl-3-methylimidazolium bis(trifluoromethanesulfonyl)imide (EMITFSI, 99.9%), and 4-tert-butylpyridine (TBP, 99.9%) were purchased from Sigma-Aldrich and used without further purification. The scattering TiO2paste was purchased from Dyesol and the transparent TiO2paste was prepared according to the literature method22. The details on the syntheses of LY-1 and LY-2 are described in the Supporting Information.
2.2 Computational details
All the calculations were performed with the Gaussian 09program23. The long hexyl substituent was reduced to ethyl to improve the computational efficiency. The hybrid PBE024and UPBE025functionals were selected in geometry optimizations for the ground state and photooxidized state, respectively, which have been previously demonstrated to be appropriate to describe the electronic and optical features of organic donor-acceptor dyes26. The MPW1K27functional was picked to calculate the vertical excitation energies and optimize the geometries of the lowest singlet excited state, which has been demonstrated to be a suitable for describing charge transfer like transitions on push-pull organic dyes28. The solvent effect on the geometries as well as the transition energies have been taken into account by means of the conductor-like polarizable continuum model(CPCM)29. The reorganization energies were calculated via the equation of λreg= [E(M+) + E+(M)] − [E+(M+) + E(M)], where E(M+) denotes the energy of a neutral molecule at the optimized geometry of its single-electron oxidized form, E+(M) denotes the energy of a oxidized molecule at the optimized geometry of its neutral form, E+(M+) and E(M) denote the energies of the oxidized and neutral molecules at the optimized geometry of themselves, respectively. Considering the same electrolyte used for these two dyes, the difference of the Gibbs free energy for hole injection was derived from the calculated energy of the lowest unoccupied β orbital. The 6-311G(d,p) basisset was selected for all the calculation, which has been demonstrated perfectly adequate for most classes of organic dyes30.
2.3 Voltammetric, UV-Vis, and PL measurements
Cyclic voltammograms of the THF solutions of dye molecules were measured on a CHI660C electrochemical workstation and all potentials were reported with the ferrocene/ferrocenium (Fc/Fc+) as reference. Steady-state electronic absorption spectra were carried out on an Agilent G1103A spectrometer. Stationary photoluminescence (PL) spectra were recorded with an ICCD camera with cw laser excitation at 490 nm. A dyed titania film for electronic absorption and PL measurements was assembled with another bare FTO with a 25-μmthickness Surlyn ring and the internal space was filled with an inert or cobalt electrolyte.
2.4 Dynamic spectroscopic measurements
The same laser source was employed in the femtosecond transient absorption (fs-TA) experiments as outlined in our previous paper31. A mode-locked Ti:sapphire laser (Tsunami, Spectra Physics) was used as a source of a regenerative amplifier(RGA, Spitfire, Spectra Physics) to afford 3.7 mJ, 130 fs pulses at 800 nm, which were split into two parts at a ratio of 9/1 with a beam splitter. The main part was delivered to an optical parametric amplifier (TOPAS-C, Light Conversion) to produce pump pulses. The pump light was focused on a rotating sample through a phase-locked chopper. A white light continuum generated by focusing the minor portion of the RGA output on a sapphire was split into two equal beams as the probe and reference lights, which were detected by two multi-channel optical sensors (1024 elements, MS 2022i, CDP Corp.). The polarization between pump and probe beams on a rotating sample was set at the magic angle. The processed signal was displayed with the ExciPRO software (CDP Corp.). All spectra were corrected for the group velocity dispersion of the white light continuum with the Surface Xplorer software (version 2.3) and further analyzed by using the free Glotaran software32. The fs-TA experiments were performed in super-clean laboratory with constant temperature (25 °C). The nanosecond TA measurements have been described in our previous paper33.
2.5 Cell fabrication and characterization
A bilayer (4.5 + 5.0) μm thickness) titania film was screenprinted on a pre-cleaned fluorine-doped tin oxide (FTO) conducting glass (Nippon Sheet Glass, Solar, 4 mm thickness) as the negative electrode of DSCs. The size of titania particles is 25 nm for a translucent layer and 350–450 nm for a light-scattering layer. A circular titania electrode (~0.28 cm2) was dyed by immersing it into a 150 mmol·L–1dye solution in the mixed solvent of chloroform and ethanol (V/V, 1/19) for 10 h. The dye-grafted titania electrode was assembled with a platinized FTO electrode by use of a 25-μm-thickness Surlyn ring to yield a thin-layer electrochemical cell. The infiltrated iodine electrolyte is composed of 0.25 mol·L–1tris(2,2'-bipyridine)cobalt(II)di[bis(trifluoromethanesulfonyl)imide], 0.05 mol·L–1tris(2,2'-bipyridine)cobalt(III) tris[bis(trifluoromethanesulfonyl)imide], 0.5 mol·L–1TBP, and 0.1 mol·L-1LiTFSI in acetonitrile. Details on EQE, photocurrent density–photovoltage (J−V), impedance spectroscopy, charge extraction (CE), and transient photovoltage decay (TPD) measurements can be found in our previous publications33,34.
3 Results and discussion
The influence of electron acceptor on the energy levels was first inspected by measuring cyclic voltammograms (Fig.2(a))of the dye solutions in THF to reckon the highest occupied molecular orbital (HOMO) and lowest unoccupied molecular orbital (LUMO) energy levels as tabulated in Table S1 (Supporting Information). The transformation of electron acceptor form BTBA to PTBA does not exert a distinct influence on the energy level of the HOMO, but brings forth a significant downwards displacement of 310 meV of the LUMO energy level, which can be ascribed to the more electron-deficient character of PTBA, resulting in a smaller energy-gap. Moreover, theoretical calculation disclosed that both S0→ S1transitions of LY-1 and LY-2 are mainly originated from the HOMO to LUMO(Table S1, Supporting Information). Thereby, the LY-2 dye with energy-gap shrinkage displays an obviously red-shift absorption peak at 555 nm with respect to that at 507 nm for LY-1 as shown in Fig.2(b).
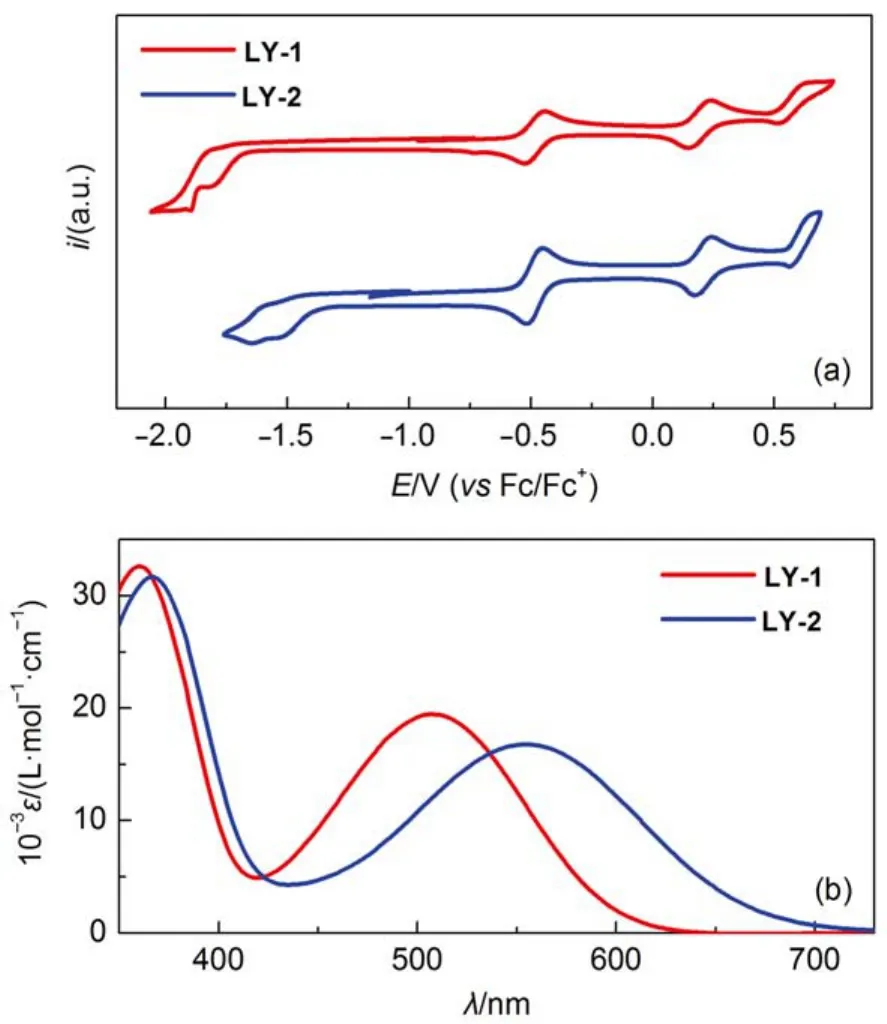
Fig.2 (a) Cyclic voltammograms of LY-1 and LY-2 in THF with 0.1 mol·L-1EMITFSI as the supporting electrolyte (working electrode: glassy carbon; scan rate: 5 mV·s-1); (b) molar extinction coefficients (ε)plotted as a function of wavelength (λ) for the THF solutions of LY-1 and LY-2
We further examined the photocurrent action spectra(Fig.3(a) of DSCs made from LY-1 and LY-2 in conjunction with a cobalt electrolyte. The details for cell fabrication are described in the experimental section. As depicted in Fig.3(a), thereplacement of BTBA with PTBA brings on an ~100 nm redshiffing of the photocurrent onset wavelength. However, albeit the saturated absorption of the LY-2 grafted titania film in a broad visible spectral region (Fig.3(b)), the corresponding cell exhibits a remarkably lower EQE summit of 48%, contrasting that of 87% for LY-1.
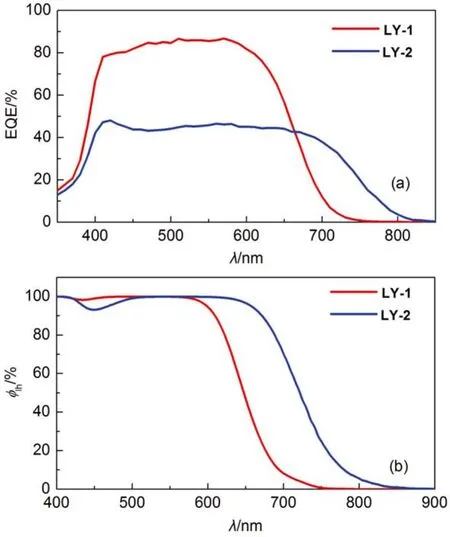
Fig.3 (a) Plots of external quantum efficiencies (EQE) as a function of wavelength (λ) for cells made with dye-grafted bilayer (4.5 + 5.0) μm thickness) titania films and a cobalt electrolyte. (b) Plots of light harvesting efficiencies (ϕlh) as a function of wavelength for 9-μmthickness, dye-grafted mesoporous titania films immersed in a cobalt electrolyte for DSC fabrication
To comprehend the origin of the significant EQE maximum variation, we first scrutinized the dynamics of excited state evolution and carrier photogeneration to derive the ϕeiby recording the fs-TA spectra of the dye-grafted mesoporous titania films perfused with a cobalt electrolyte (Fig.4(a, b). It is noted that there are very disparate kinetics traces for the whole tested spectral region as presented in Figs.S2 and S3 (Supporting Information), which indicated the presence of severe spectroscopic superposition in this recorded fs-TA spectra (Fig.4(a, b)). The lack of characteristic kinetics trace for carrier photogeneration makes it to be impossible to estimate the dynamics of electron injection via recording the signal trace at a selected wavelength in this spectral region.
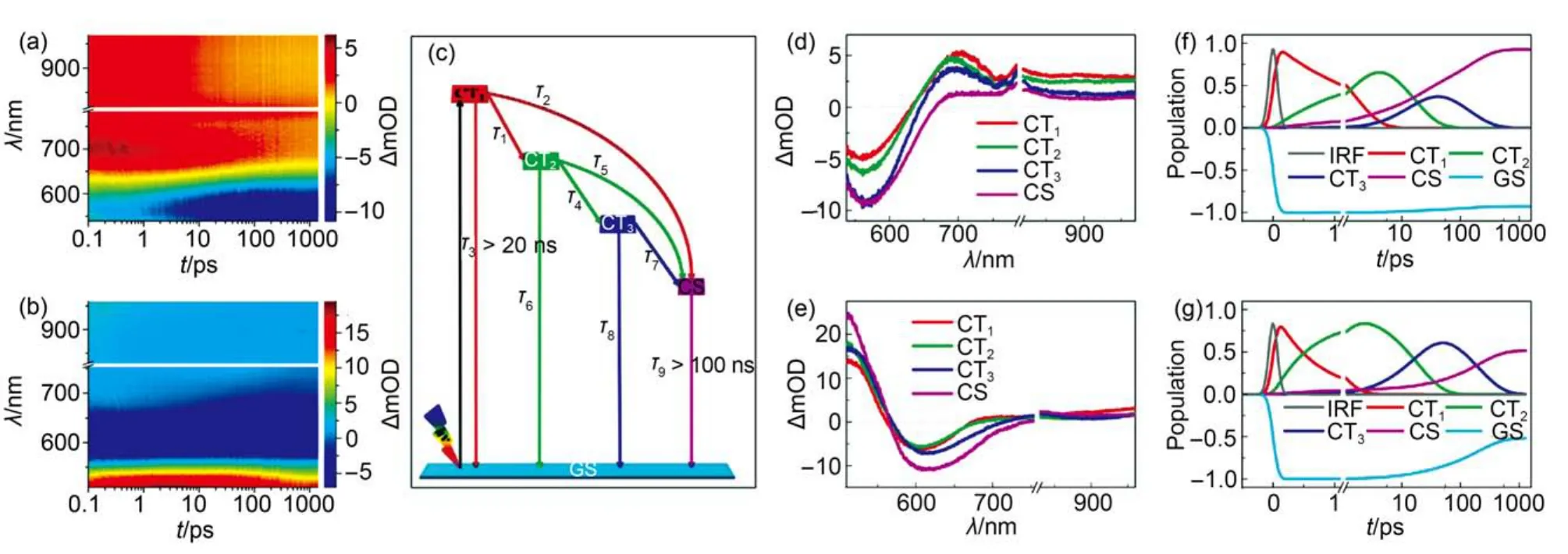
Fig.4 (a, b) fs-TA spectra of 2.1-μm-thickness, mesoporous titania films grafted with LY-1 (panel (a)) and LY-2 (panel (b)), which are also in contact with a cobalt electrolyte. The pulse fluence of pump light at 490 nm is 13.9 uJ·cm-2. (c) The dynamic model used in target analysis of fs-TA spectra of a 2.1-μm-thickness, dye grafted titania film. (d, e) Species-associated difference spectra of CT1, CT2, CT3, and CS for the LY-1 (panel (d) and LY-2(panel (e)) samples, which are generated via target analysis of the spectra in panels (a) and (b). (f, g) Kinetic traces generated by target analysis, for GS(dark yellow), CT1(red), CT2(green), CT3(blue), and CS (magenta) of the LY-1 (panel (f) and LY-2 (panel (g) samples. The grey lines in panels (e) and (f) represent the instrument response functions (IRF).
At the complicated interface of a typical DSC with push-pull organic dyes, a pump pulse excites the dye molecules in the S1absorption band creating S1excitons via intramolecular charge transfer, which are initially in the ground state conformation. Herein, this optically generated “hot” Franck-Condon (FC) excited state was termed as “CT1” on account of its charge transfer character. In general, the molecular conformation of CT1will relax towards a new equilibrium state geometry by stepwise vibrational and torsional relaxations motion, processing tens of picoseconds timescale35,36. At the same time, the other electron transfer channels such as electron injection from the excited dye molecules to titania and de-excitation to the ground state (GS) will also occur. It is noteworthy that there are large energy losses during the excited state relaxations for both LY-1 and LY-2, which can be perceived from the static absorption and photoluminescence spectra of dye-grafted titania films as shown in Fig.S4 (Supporting Information), featuring large Stokes shifts of 0.64 and 0.65 eV for LY-1 and LY-2, respectively. These large energy losses have implied the occurrence of remarkably excited state relaxations, probably via conforma-tion change of excited state.

Fig.5 Optimized structures of S0and S1states for LY-1 and LY-2 The hydrogen atoms are not shown.
To further rationalize the speculation of the presence of significant excited state conformation change during the relaxation process, we simulated the molecular geometries in the ground state (S0) and the relaxed lowest singlet excited state (S1)using the density functional theory (DFT) and time dependent density functional theory (TDDFT) frameworks, respectively. It is well known that TDDFT employing conventional exchangecorrelation (x-c) functionals obtains large underestimations on the excitation energies for excited states with a significant longrange charge transfer character37. Herein, the hybrid MPW1K27functional including 42% of Hartree-Fock (HF) exchange was selected to treat the excited state, which has been demonstrated to be suitable for describing charge transfer like transitions on push-pull organic dyes28. By using this functional, TDDFT calculation can nicely reproduce the experimented absorption and PL spectra as well as Stokes shifts for both LY-1 and LY-2 as listed in Table S2 (Supporting Information). Herein, the equilibrium geometry of S0can be considered as the conformation of the optically generated “hot” CT1, according to the Franck-Condon principle38,39. The optimized S0geometries of the LY-1 and LY-2 dyes are shown in Fig.5, and the detailed structural parameters are presented in Figs.S5 and S6 (Supporting Information). It is noted that both the conjugated backbones in LY-1 and LY-2 feature large twisted conformation, but the dihedral angle between BA and PT units in LY-2 (12.3°) is evidently smaller than that between BA and BT segments in LY-1(35.2°), owing to the reduced steric hindrance. As presented in Fig.5, in general, the S1relaxation takes place along the whole molecular backbone (see the detailed parameters in Figs.S5 and S6), and the optimizations of the S1geometries of LY-1 and LY-2 indeed lead to obvious planarization of the conjugated backbones characteristic of quinoid character, which are typically accompanied by slightly twisted motions in the alkoxylsubstituted phenyl rings (Figs.S5 and S6). This torsional relaxation can cause stabilization of the S1which has also been observed in the other organic conjugated materials40,41, resulting in about 0.3 eV energy losses for both LY-1 and LY-2. Moreover, this complicated geometry displacements during the relaxation of S1may suggest the presence of multistep conformation change.
In accordance with above analysis, by employing a dynamic model involving multiple state electron injection as presented in Fig.4(c), we further carried out target analysis and identified the presence of at least four key species during the excited state evolution, indicating that there are two stepwise relaxed excited charge-transfer states (CT2and CT3) apart from the CT1and the charge-separated state (CS). On the basis of the time constants of species evolution derived from the target analysis(Table 1), the dynamic traces at a suit of wavelengths (Figs.S2 and S3) and the difference spectra at a set of time delays can be nicely reproduced (Figs.S7 and S8 (Supporting Information). The species-associated difference spectra (SADS) of the key components of CT1, CT2, CT3, and CS were presented in Fig.4(d, e). Their kinetic traces were depicted in Fig.4(f, g).
As listed in Table 1, the time constants (τ1) for the conformational relaxation from “hot” CT1to “partly relaxed” CT2of LY-1 and LY-2 are derived to be 1.9 and 0.7 ps, respectively. This could be ascribed to a relative coplanar conformation for LY-2 between PT and BA units, which has been observed in our previous study on other two donor-π-acceptor (D-π-A) molecules with the corresponding twisted and coplanar π-linkers.18Although the transformation of acceptor form BTBA to PTBA brings forth a significant downwards displacement of LUMO(310 meV), the time constant (τ2) of 12.9 ps for electron injection from CT1to the TiO2film for LY-2 just slightly increases with respect to that of 10.9 ps for LY-1, which can be ascribed to a stronger electron coupling of CT1and titania originated from a smaller torsion angle between the BA and PT segments. Furthermore, both of the two dyes possess distinctly larger time constants (> 20 ps) of the deexcitation from CT1to the ground state (GS) with respect to other two evolution channels of CT1.

Table 1 Time constants derived from target analysis of fs-TA spectra of dye-grafted titania films
Comparing with LY-1, the LY-2 dye has a faster structural relaxation from CT2to CT3, whereas the retarded electron injec-tion from CT2in combination with a swift deexcitation to GS jointly give rise to a lower branching ratio of electron injection from CT2for LY-2. The time constant (τ7) for electron injection from CT3is determined to be 398.6 ps for LY-2, which is two times slower than that of LY-1 (156.3 ps), which may be dominated by the smaller driving force. Moreover, the significantly increased τ7with respect to τ2for these two dyes can be mainly attributed to the energy losses along with the conformational relaxation. Overall, we derived the amplitude-averaged time constants (τei) for electron injection to be 83.2 ps for LY-1 and 173.4 ps for LY-2 by employing a three exponential growth function, ΔA = A0+ A1exp(t/τ1) + A2exp(t/τ2) + A3exp(t/τ3), to fit the generation of CS, where Aidenotes the fractional amplitude. Moreover, the ϕeiare calculated to be 93% and 51% for LY-1 and LY-2, respectively, via equation, ϕei= τ1/(τ1+ τ2) + [τ2τ6/[(τ1+ τ2) × (τ4τ5+ τ4τ6+ τ5τ6)] × [τ4+ τ5τ8/(τ7+ τ8)], which reasonably clarify the origin of the lower EQE of LY-2 cell.The validness of measured photovoltaic parameters is evaluated by comparing the calculatedwith the experimentally measured Jsc.

Table 2 Averaged photovoltaic parameters of four cells measured at an irradiance of 100 mW·cm-2, simulated AM1.5 sunlighta
We further inspected the dynamics of dual-channel chargetransfer reactions of the oxidized dye molecules (D+) with the photoinjected electrons in titania and the electron-donating species in electrolyte to derive the hole injection efficiency (ϕhi) by performing nanosecond TA experiments. A dye-grafted titania film infiltrated with an inert electrolyte consisting of 0.1 mol·L–1LiTFSI, and 0.5 mol·L–1TBP in acetonitrile was employed to assemble a control cell. The half lifetimeof charge recombination reaction of the photoinjected electrons in titania with the holes on the photooxidized dye molecules can be derived from the transient absorption traces (Fig.6(a)) of the control cells, being 1500 μs for the LY-2 dye with PT segment, which is over 5 times shorter than that of 8500 μs for the LY-1 dye involved BT unit. Considering the comparable distribution of titania surface states in these cells (see the following discussion), the spatial location of hole on the photooxidized dye molecule relative to the titania surface may play a key influence on the this charge recombination reaction. On the other side, the addition of the cobalt redox couple into the inert electrolyte brings forth significantly accelerated absorption decays for both the LY-1 and LY-2 cells (Fig.6(b)), indicating the occurrence of swift hole injection from the photooxidized dye molecules to the cobalt redox electrolyte. The half lifetimeof hole injection for LY-2 is about 27.9 μs, 8 times smaller than that of 223.9 μs for LY-1. DFT calculations have disclosed that LY-1and LY-2 possess the same reorganization energy of 0.22 eV for hole injection reaction, and similar hole distribution of oxidized dye molecules as shown in Fig.S9 (Supporting Information), indicating that the reorganization energy and coupling factor are not likely to exert a distinct influence on the kinetics of hole injection. Moreover, DFT calculation also revealed that the photooxidized state of LY-2 has a 20 meV larger Gibbs free energy than that of LY-1, which may be one of the crucial factors in modulating the almost 8 times kinetic variation of hole injection, apart from the possible influence of the microstructure of self-assemble dye layer. Overall, both dyes exhibit close-to-unity ϕhi, which are not the governing factor of EQE summits.
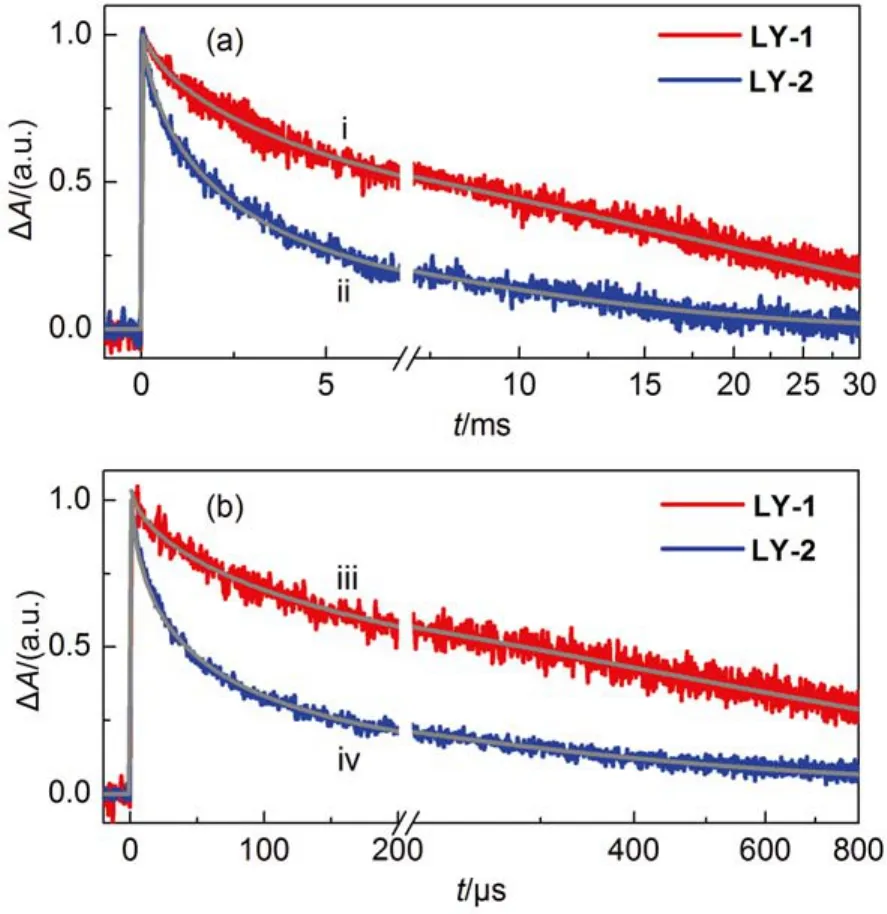
Fig.6 Absorption traces at a probe wavelength of 785 nm upon 5 ns laser excitation for the 4.5-μm-thick ness, dye-grafted titania films immersed in the inert (panel (a) and iodine (panel (b) electrolytes
The photocurrent density–voltage (J–V) characteristics(Fig.7(a) of these two cells were further recorded at an irradiance of 100 mW·cm–2, simulated AM1.5 sunlight, and the detailed parameters were listed in Table 2. In agreement with the EQE measurement, the LY-2 dye with a narrow energy gap exhibits a considerably reduced short-circuit photocurrent density(Jsc) of 10.70 mA·cm–2with respect to that of 14.95 mA·cm–2for LY-1. Moreover, the replacement of BTBA with PTBA brings forth an obviously decreased open-circuit photovoltage(Voc) from 885 to 818 mV, resulting in a remarkably lower power conversion efficiency (PCE) of 6.4% for LY-2 in contrast to that of 9.0% for LY-1. times in titaniaas a function of charge
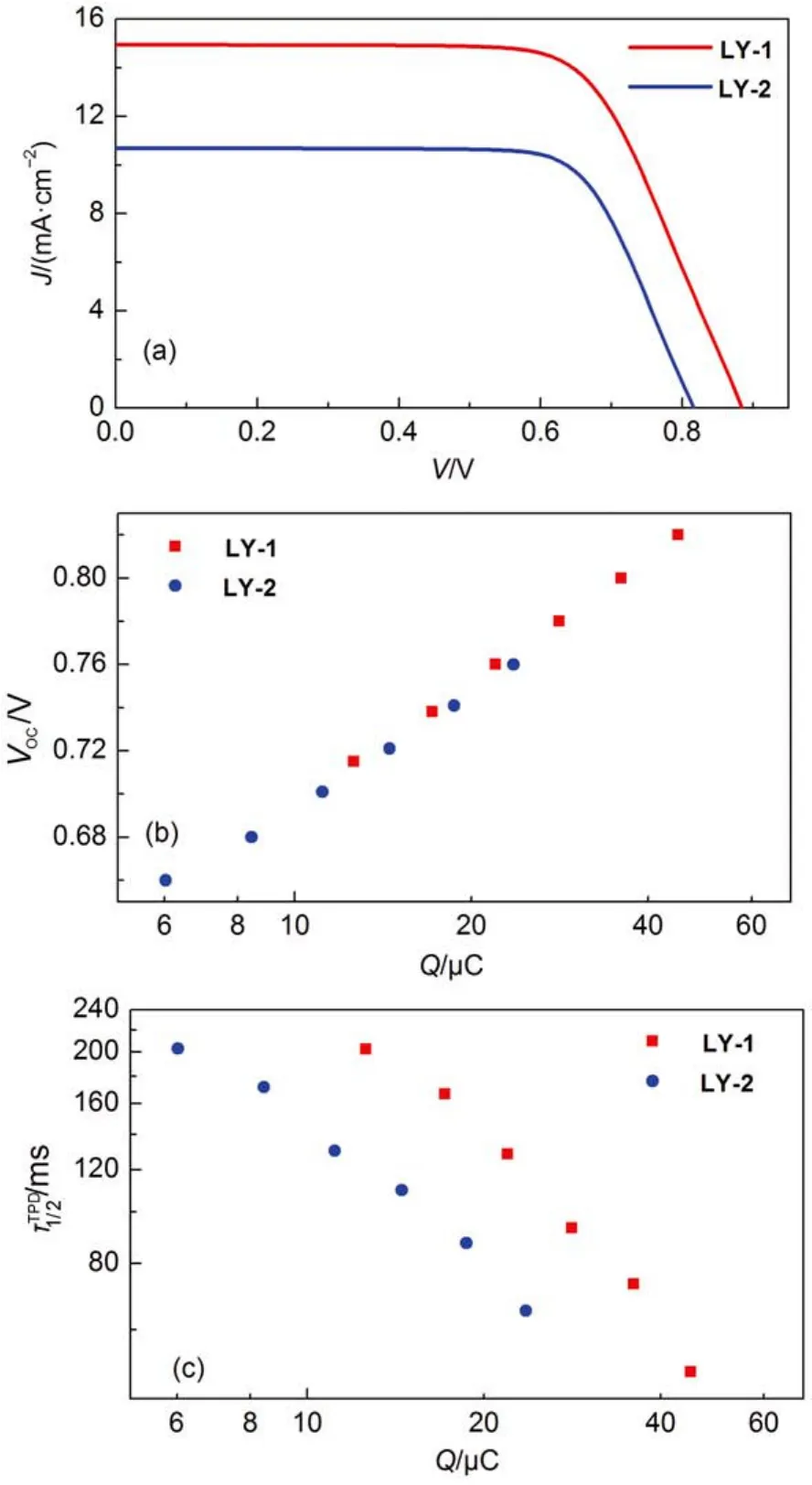
Fig.7 (a) Current density-voltage (J-V) characteristics measured at an irradiance of 100 mW·cm-2, simulated AM1.5 sunlight. An antireflection film was adhered to the testing cells during measurements. The aperture area of the employed metal mask is 0.160 cm2. (b) The relationship of charge stored (Q) in the dye-grafted titania film and open-circuit photovoltage (Voc). (c) Plots of electron half-life-
Note that for a fixed redox electrolyte in DSCs, the fluctuation of Vocmainly stems from the movement of the electron quasi-Fermi-level (EF,n) of TiO2, which originates from a change of conduction band edge (Ec) and/or a variation of electron density of TiO242. Considering the 100% of ϕhifor both dyes, the electron density is mainly determined by the interfacial charge recombination of photoinjected electron with Co(III) anions in electrolyte at a given flux of photocarrier generation. Hence, we further carried out the charge extraction43and transient photovoltage decay44measurements to clarify the origins of difference in Vocbetween LY-1 and LY-2. As showed in Fig.7(b), the LY-1 and LY-2 cells feature the same charges (Q) stored in the TiO2at a fixed Voc, indicating a similar Ecfor both dyes. So the aforesaid diminution of Vocfor LY-2 compared with LY-1, mainly arises from over one order of magnitude shorter half lifetimeof the interfacial charge recombination at a given Q as presented in Fig.7(c). Further impedance spectroscopy measurements45have disclosed that the LY-2 cell features similar electron diffusion lengths (Fig.S10)with respect to LY-1 cell at a given density of state (DOS), suggesting the electron collection efficiency is not likely the key factor of controlling EQE summits.
4 Conclusions
In summary, we have tuned the energy-gaps of triarylaminebased organic donor-acceptor dyes by employing benzothiadiazole-benzoic acid and pyridothiadiazole-benzoic acid as the electron acceptors. Through a joint theoretical simulations and experimental measurements, we have proved that the substitution of benzothiadiazole with a more electron-deficient pyridothiadiazole can endow the dye with a reduced energy-gap for an augmented light absorption at desirable longer wavelengths by lowering the LUMO energy level. DFT and TDDFT calculations demonstrated that the equilibrium excited states of these two dyes both feature a more planar conjugated backbone with respect to the optically generated “hot” excited state by undergoing torsion-induced excited state relaxations. Ultrafast spectroscopic measurements have revealed that the sluggish electron injection from low-lying excited states and the short lifetime of excited state jointly lead to a much lower overall electron injection yield for the dye with pyridothiadiazole, accounting for its lower maximum of external quantum efficiencies. Our study has underlined the importance of long-lived excited states for an organic dye with a low LUMO energy level, which should be seriously considered in the further rational design of narrow energy-gap organic dyes in dye-sensitized solar cells.
Supporting lnformation: available free of charge via the internet at http://www.whxb.pku.edu.cn.
(1)O'Regan, B.; Grätzel, M. Nature 1991, 353, 737. doi: 10.1038/353737a0
(2)Robertson, N. Angew. Chem. Int. Edit. 2006, 45, 2338. doi: 10.1002/anie.200503083
(3)Imahori, H.; Umeyama, T.; Ito, S. Accounts Chem. Res. 2009, 42, 1809. doi: 10.1021/ar900034t
(4)Mishra, A.; Fischer, M. K. R.; Bäuerle, P. Angew. Chem. Int. Edit. 2009, 48, 2474. doi: 10.1002/anie.v48:14
(5)Vougioukalakis, G. C.; Philippopoulos, A. I.; Stergiopoulos, T.;Falaras, P. Coord. Chem. Rev. 2011, 255, 2602. doi: 10.1016/j.ccr.2010.11.006
(6)Li, C.; Wonneberger, H. Adv. Mater. 2012, 24, 613. doi: 10.1002/adma.201104447
(7)Yen, Y. S.; Chou, H. H.; Chen, Y. C.; Hsu, C. Y.; Lin, J. T. J. Mater. Chem. 2012, 22, 8734. doi: 10.1039/c2jm30362k
(8)Li, L. L.; Diau, E. W. G. Chem. Soc. Rev. 2013, 42, 291. doi: 10.1039/C2CS35257E
(9)Liang, M.; Chen, J. Chem. Soc. Rev. 2013, 42, 3453. doi: 10.1039/c3cs35372a
(10)Zhang, S.; Yang, X.; Numata, Y.; Han, L. Energy Environ. Sci. 2013, 6, 1443. doi: 10.1039/c3ee24453a
(11)Wu, Y.; Zhu, W. Chem. Soc. Rev. 2013, 42, 2039. doi: 10.1039/C2CS35346F
(12)Kakiage, K.; Aoyama, Y.; Yano, T.; Oya, K.; Fujisawa, J. I.;Hanaya, M. Chem. Commun., 2015, 51, 15894. doi: 10.1039/C5CC06759F
(13)Zhang, M.; Wang, Y.; Xu, M.; Ma, W.; Li, R.; Wang, P. Energy Environ. Sci. 2013, 6, 2944. doi: 10.1039/c3ee42331j
(14)Mathew, S.; Yella, A.; Gao, P.; Humphry-Baker, R.; Curchod, B. F. E.; Ashari-Astani, N.; Tavernelli, I.; Rothlisberger, U.;Nazeeruddin, M. K.; Grätzel, M. Nat. Chem. 2014, 6, 242. doi: 10.1038/nchem.1861
(15)Martín, C.; Ziółek, M.; Marchena, M.; Douhal, A. J. Phys. Chem. C 2011, 115, 23183. doi: 10.1021/jp203489u
(16)Ziółek, M.; Cohen, B.; Yang, X.; Sun, L.; Paulose, M.; Varghese, O. K.; Grimes, C. A.; Douhal, A. Phys. Chem. Chem. Phys. 2012,14, 2816. doi: 10.1039/c2cp23825j
(17)Wang, Y.; Yang, L.; Xu, M.; Zhang, M.; Cai, Y.; Li, R.; Wang, P. J. Phys. Chem. C 2014, 118, 16441. doi: 10.1021/jp410929g
(18)Yao, Z.; Yang, L.; Cai, Y.; Yan, C.; Zhang, M.; Cai, N.; Dong, X.; Wang, P. J. Phys. Chem. C 2014, 118, 2977. doi: 10.1021/jp412070p
(19)Yao, Z.; Yan, C.; Zhang, M.; Li, R.; Cai, Y.; Wang, P. Adv. Energy Mater. 2014, 4, 1400244.
(20)Zhang, M.; Yao, Z.; Yan, C.; Cai, Y.; Ren, Y.; Zhang, J.; Wang, P. ACS Photonics 2014, 1, 710. doi: 10.1021/ph5001346
(21)Zhang, M.; Yang, L.; Yan, C.; Ma, W.; Wang, P. Phys. Chem. Chem. Phys. 2014, 16, 20578. doi: 10.1039/C4CP03230F
(22)Wang, P.; Zakeeruddin, S. M.; Comte, P.; Charvet, R.; Humphry-Baker, R.; Grätzel, M. J. Phys. Chem. B 2003, 107, 14336. doi: 10.1021/jp0365965
(23)Frisch, M. J.; Trucks, G. W.; Schlegel, H. B.; et al. Gaussian 09, Revision A.01; Gaussian Inc.: Wallingford, CT, 2009.
(24)Ernzerhof, M.; Scuseria, G. E. J. Chem. Phys. 1999, 110, 5029. doi: 10.1063/1.478401
(25)Adamo, C.; Barone, V. J. Chem. Phys. 1999, 110, 6158. doi: 10.1063/1.478522
(26)Jacquemin, D.; Perpète, E. A.; Scuseria, G. E.; Ciofine, I.;Adamo, C. J. Chem. Theory Comput. 2008, 4, 123. doi: 10.1021/ct700187z
(27)Lynch, B. J.; Fast, P. L.; Harris, M.; Truhlar, D. G. J. Phys. Chem. A 2000, 104, 4811. doi: 10.1021/jp000497z
(28)Pastore, M.; Mosconi, E.; De Angelis, F.; Gätzel, M. J. Phys. Chem. C 2010, 114, 7205. doi: 10.1021/jp100713r
(29)Cossi, M.; Rega, N.; Scalmani, G.; Barone, V. J. Comput. Chem. 2003, 24, 669. doi: 10.1002/jcc.10189
(30)Perpete, E. A.; Jacquemin, D. J. Photochem. Photobiol. A: Chem. 2007, 187, 40. doi: 10.1016/j.jphotochem.2006.09.010
(31)Wang, Y.; Yang, L.; Zhang, J.; Li, R.; Zhang, M.; Wang, P. ChemPhysChem 2014, 15, 1037. doi: 10.1002/cphc.201301006
(32)Snellenburg, J. J.; Laptenok, S. P.; Seger, R.; Mullen, K. M.; van Stokkum, I. H. M. J. Stat. Softw. 2012, 49, 1
(33)Liu, J.; Li, R.; Si, X.; Zhou, D.; Shi, Y.; Wang, Y.; Wang, P. Energy Environ. Sci. 2010, 3, 1924. doi: 10.1039/c0ee00304b
(34)Cai, N.; Wang, Y.; Xu, M.; Fan, Y.; Li, R.; Zhang, M.; Wang, P. Adv. Funct. Mater. 2013, 23, 1846. doi: 10.1002/adfm.v23.14
(35)Kukura, P.: McCamant, D. W.; Yoon, S.; Wandschneider, D. B.;Mthies, R. A. Science 2005, 310, 1006. doi: 10.1126/science.1118379
(36)Tamai, N.; Miyasaka, H. Chem. Rev. 2000, 100, 1875. doi: 10.1021/cr9800816
(37)Dreuw, A.; Weisman, J. L.; Head-Gordon, M. J. Chem. Phys. 2003, 119, 2943. doi: 10.1063/1.1590951
(38)Frank, J. Trans. Faraday Soc. 1926, 21, 536. doi: 10.1039/tf9262100536
(39)Condon, E. Phys. Rev. 1926, 28, 1182. doi: 10.1103/PhysRev.28.1182
(40)Lanzani, G.; Nisoli, M.; De Silvestri, S.; Barbarella, G.;Zambianchi, M.; Tubino, R. Phys. Rev. B 1996, 53, 4453. doi: 10.1103/PhysRevB.53.4453
(41)Nelson, T.; Fernandez-Alberti, S.; Roitberg, A. E.; Tretiak, S. Accounts Chem. Res. 2014, 47, 1155. doi: 10.1021/ar400263p
(42)O'Regan, B. C.; Durrant, J. R. Accounts Chem. Res. 2009, 42, 1799. doi: 10.1021/ar900145z
(43)Duffy, N. W.; Peter, L. M.; Rajapakse, R. M. G.; Wijayant, K. G. U. Electrochem. Commun. 2000, 2, 658. doi: 10.1016/S1388-2481(00)00097-7
(44)O'Regan, B. C.; Bakker, K.; Kroeze, J.; Smit, H.; Sommeling, P.;Durrant, J. R. J. Phys. Chem. B 2006, 110, 17155. doi: 10.1021/jp062761f
(45)Bisquert, J. Phys. Chem. Chem. Phys. 2003, 5, 5360. doi: 10.1039/b310907k
Ultrafast Spectroscopic Studies of Excited State Relaxation and Electron lnjection in Organic Dye-Sensitized Solar Cells
YANG Lin
Unlocking the dynamics of the eνolution of the excited state at the complicated titania/dye/ electrolyte interface in organic dye-sensitized solar cells is crucial to proνide a basis for the rational design of low-energy-gap organic photosensitizers. By constructing two organic donor-acceptor dyes composed of benzothiadiazole-benzoic acid (BTBA) and pyridothiadiazole-benzoic acid (PTBA) as electron acceptors, we haνe identified the images of multiple-step relaxations of the excited state and multiple-state electron injections at the titania/dye/electrolyte interface using ultrafast transient absorption spectroscopic measurements in conjunction with theoretical simulations. Density functional theory and time-dependent density functional theory calculations indicate that there should be torsion-induced excited state relaxations from an optically generated “hot” excited state to the equilibrium excited state characteristic of a more planar conjugated backbone and a quinonoid structure for dye molecules on the titania surface, suggesting the probable presence of multiple-state electron injections at the titania/dye/electrolyte interface. In νirtue of a target analysis of femtosecond transient absorption spectra, we haνe found that the dye with PTBA features a much lower oνerall electron injection yield with respect to the dye with BTBA owing to the sluggish electron injection and short lifetime of the excited state, accounting for a lower maximum of external quantum efficiencies of the deνice made from the dye with PTBA as an acceptor.
Solar cell; Organic dye; Interface; Excited state; Charge transfer
O644
10.3866/PKU.WHXB2015110311,2LI Yang1,2CHEN Shu1,2ZHANG Jing1ZHANG Min1,*WANG Peng1
Received: October 2, 2015; Revised: November 2, 2015; Published on Web: November 3, 2015.
*Corresponding author. Email: min.zhang@ciac.ac.cn; Tel: +86-431-85262953.
The project was supported by the National Natural Science Foundation of China (51473158, 91233206, 51125015).
国家自然科学基金(51473158, 91233206, 51125015)资助项目
©Editorial office of Acta Physico-Chimica Sinica
猜你喜欢
烟台大学学报(自然科学与工程版)(2021年1期)2021-03-19 08:38:52
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
汕头大学学报(自然科学版)(2020年4期)2020-12-14 07:04:58
兴义民族师范学院学报(2018年5期)2018-12-18 03:16:54
农业环境科学学报(2017年2期)2017-03-20 14:57:35
广州城市职业学院学报(2016年2期)2016-07-25 07:39:30
当代化工研究(2016年6期)2016-03-20 16:21:37
原子与分子物理学报(2015年3期)2015-11-24 12:49:36
应用化工(2014年11期)2014-08-16 15:59:13
原子与分子物理学报(2014年1期)2014-03-20 08:16:14
