
LA/C14DMAO/H2O体系多结构自组装体及其模板金纳米材料制备与性能
2016-11-18 07:29:27郝京诚
物理化学学报 2016年1期
冯 磊 郝京诚
(山东大学,胶体与界面化学教育部重点实验室,济南 250100)
LA/C14DMAO/H2O体系多结构自组装体及其模板金纳米材料制备与性能
冯 磊 郝京诚*
(山东大学,胶体与界面化学教育部重点实验室,济南 250100)
月桂酸(LA)与十四烷基二甲基氧化胺(C14DMAO)形成的无盐阴/阳离子表面活性剂混合体系表现出丰富的相行为。运用冷冻蚀刻透射电子显微镜 (FF-TEM)和偏光显微镜(POM)、差示扫描量热(DSC)、流变和2H NMR测定对体系相行为和微观结构进行了研究,发现水溶液中可自聚集形成胶束(L1)、层状(Lαl)、囊泡(Lαν)和凝胶相。以胶束相和层状相为软模板制备了金纳米材料,运用透射电子显微镜(TEM)和能谱仪(EDS)表征了金纳米材料。与用传统阳离子表面活性剂溶液制备金纳米材料相比,该体系由于具有自身还原性而不需要加入还原剂NaBH4。实验证明:还原过程不会破坏模板溶液原有微观结构,且可通过调控聚集体结构实现控制制备金纳米材料形貌的目的。HK-2细胞的噻唑蓝(MTT)比色法实验进一步证明,本体系制备的球形金纳米材料作为基因载体具有高效和低毒的特点,在基因治疗中具有潜在的实际应用价值,可为寻求安全可靠的基因治疗途径提供实验数据和理论参考。
月桂酸;十四烷基二甲基氧化胺;相行为;模板;金纳米材料
1 引 言
众所周知,表面活性剂混合体系相较于单一表面活性剂体系更易自组装形成各种形态的聚集体,且混合体系表现出多样的相行为和溶液性质。例如,混合体系通常比单一组分具有更低的表面张力和临界胶束浓度;由于阴、阳离子表面活性剂可通过静电作用形成离子对,它们在水溶液中可形成多结构的聚集体。自1989年美国科学家Kaler等1观察到阴/阳离子表面活性剂复配体系中自发形成囊泡以来,不同领域科学家对该类体系进行了深入系统的研究。阴/阳离子表面活性剂混合体系包括有盐及无盐两类。早期研究主要集中在含盐体系,这类体系中,由于溶液中表面活性剂反离子形成盐对膜电荷有静电屏蔽作用,在等摩尔或接近等摩尔混合时产生大量的沉淀,限制了其应用2–5。而无盐阴/阳离子表面活性剂溶液体系由于其低离子强度、低渗透压以及聚集体双分子层膜表面电荷未被静电屏蔽等优点,可以形成具有独特结构及性质的聚集体,逐渐成为关注的热点6–11。
金纳米材料由于在催化、传感、生物成像、药物传输等方面所体现出的优异电学、光学及热学性质,其形貌和尺寸的可控制备也受到了国内外学者的广泛重视12–22。本文研究了月桂酸(LA)与十四烷基二甲基氧化铵(C14DMAO)所构筑的无盐阴/阳离子表面活性剂混合体系的相行为及两者在水溶液中的聚集特性。烷基二甲基氧化胺是一种非离子型表面活性剂,可以通过调控溶液pH值改变其质子化程度。在酸性条件下,C14DMAO由于质子化,结合H+而使胶束带正电荷,溶液表现出阳离子表面活性剂的性质;而在中性和碱性条件下则表现出非离子表面活性剂性质。在本体系中,我们选择胶束(L1相)和层状(Lαl相)溶液作为软模板,利用质子化后C14DMAOH+的羟基所具有的还原性,实现了在溶液中通过C14DMAOH+离子还原氯金酸(HAuCl4)制备了金纳米材料。与传统阳离子表面活性剂溶液制备金纳米材料相比,本方法无需加入NaBH4、抗坏血酸、柠檬酸钠等还原剂,样品溶液既作为模板溶液,又兼具还原性。实验证明,还原过程不会破坏模板溶液原有微观结构,并可通过调控模板溶液的自组装结构以实现金纳米材料形貌的可控制备23,提供了纳米材料制备的新途径。纳米材料作为基因载体应用于基因治疗领域的研究,目前主要集中在阳离子高分子纳米材料,如聚乙烯亚胺、多聚碱性氨基酸等24–26。本实验中我们将合成的金纳米材料作为基因载体,对其潜力进行了初步研究。相较于传统表面活性剂制备的金纳米材料,本文合成的金纳米材料可以在更低浓度下压缩DNA分子,并且通过人肾小管上皮细胞HK-2细胞的噻唑蓝(MTT)比色法实验证明,其具有更好的生物兼容性。本文的研究结果为寻求安全可靠的基因治疗途径提供理论参考,并对药物缓释、基因传递、无机纳米材料的软模板制备等具有指导意义。
2 材料与方法
2.1 实验试剂
十四烷基二甲基氧化胺由Clariant公司提供的25%(质量分数,w)的溶液,干燥后经丙酮两次重结晶得到;月桂酸(LA),化学纯,梯希爱上海化成试剂有限公司;氯金酸(HAuCl4),化学纯,西亚试剂;双链DNA钠盐(dsDNA),500–600 碱基对,ACROS(美国);噻唑蓝,纯度≥ 98%, Amresco(美国);胎牛血清,杭州四季青生物工程材料有限公司;青霉素和链霉素,北京索莱宝科技有限公司;人肾小管上皮细胞 HK-2,上海沪震生物科技有限公司;实验中所用超纯水由成都优普UPHWIII-90T型超纯水仪制备,其电阻率为18.25 MΩ·cm。
2.2 样品制备
样品配制及相行为研究:在C14DMAO的浓度(cC14DMAO) < 100 mmol·L–1时,将120个LA与C14DMAO的混合样品溶液在(25.0 ± 0.1) °C下平衡四周以上,期间不断振荡样品加快其相平衡,直至观察溶液的外观不再发生变化。相边界的确定通过目视和偏光观察,并辅以电导率测定。
金纳米颗粒的制备:配制 20 mmol·L–1的氯金酸(HAuCl4)溶液,避光低温保存。将HAuCl4溶液缓慢滴加到低速搅拌的模板溶液中,滴加完成后,避光继续低速搅拌12 h。随后,以8000 r·min–1离心模板溶液30 min,保留离心管底部沉淀,倾掉模板溶液,加入超纯水,涡旋振荡,离心分离,重复三次。将清洗干净的金纳米材料放入干燥器中室温干燥,密封保存。
2.3 实验仪器和方法
电导率及pH值的测定:使用上海雷磁仪器厂生产的型号为DDSJ-308F的电导率仪和型号为DJS-10C的电极在(25.0 ± 0.5) °C下测定溶液电导率。使用德国赛多利斯公司生产的PB-21 pH计在(25.0 ± 0.5) °C下测定pH值。
流变测定:流变数据的测定在美国HAAKE 公司RS6000流变仪上通过锥-平板转子系统(C35/1)测量,频率扫描的范围为0.01–10 Hz。所有样品在测试之前均恒温平衡稳定至少四周。
冷冻蚀刻透射电镜观察(FF-TEM):样品的刻蚀和复形通过德国Leica公司EM BAF 060冷冻蚀刻制样台完成,通过日本JEM-1400电子显微镜(JEOL)观察,电压120 kV,图像使用美国Gatan 公司831 CCD仪(2.7 K × 2.7 K)采集记录。
偏光观察是在德国ZEISS公司AXIOSKOP 40/40 FL显微镜上进行,将几滴溶液滴在载玻片上,用盖玻片盖住以免溶液挥发。
氘核磁共振(2H NMR)波谱表征:样品用重水(D2O)配制(1.0 mL),平衡两周后转移至核磁管(直径为5 mm),继续平衡两周。使用配置脉冲场梯度模块(z-轴)的德国Bruker公司Avance 400核磁仪进行2H NMR测定。
金纳米材料的表征:将放置金纳米材料的离心管中加入适量的乙醇,超声30 min,然后将乙醇分散滴加在铜网上,在红外灯下烘干铜网,在日本JEM-1400电子显微镜下观察,电压120 kV,图像通过美国Gatan公司831 CCD仪(2.7 K × 2.7 K)采集记录,元素分析结果通过英国OXFORD公司INCA-X能谱仪采集。
金纳米材料与DNA分子相互作用:用紫外-可见(UV-Vis)光谱表征,使用带有变温装置的日本日立公司U-4100紫外分光光度计,纯水作为空白,测试光路长度为1 cm,波长扫描范围为400–700 nm和220–320 nm。样品溶液用一系列特定浓度金纳米材料储备液与DNA储备液混合配制而成。样品溶液在振荡培养箱中振荡1 h,静置12 h后观察变化。
细胞毒性实验(MTT法):人肾小管上皮细胞HK-2使用加有10%胎牛血清(FBS)、青霉素(100 units·mL–1)、链霉素(100 µg·mL–1)的RPMI-1640培养液在5%CO2培养箱中37 °C下培养48 h。将培养好的细胞消化、离心、重悬后计数,调整细胞浓度为1 × 104个/mL,以每孔体积200 µL接种于96孔培养板,各物质每一染毒浓度均设3个复孔。细胞染毒培养24 h后,取出培养板, 每孔加入20 µL MTT溶液, 放回培养箱继续孵育4 h。然后每孔加入150 µL 二甲基亚砜(DMSO),取上层清液使用美国Thermo公司生产的型号为Multiskan FC酶标仪于490 nm波长测定各孔吸光度(OD),取三次平行实验结果计算得出。
3 结果与讨论
3.1 LA/C14DMAO/H2O体系相行为
由于LA在水中溶解度很小,所以LA/C14DMAO/ H2O体系是C14DMAO胶束水溶液中增溶LA分子,然后二者相互作用形成各种聚集结构。首先将不同量的LA加入到一系列固定浓度的C14DMAO水溶液中,振荡混合均匀之后置于(25.0 ± 0.1) °C条件下平衡四周以上。观察样品溶液并辅以电导率等手段确定体系相边界,绘出相图,如图1a所示。混合体系表现出丰富的相行为:均匀的胶束(L1相);上部为浅蓝色的Lα相而下部为透明L1相两相;粘度较低可以流动的Lα相,粘度很高的凝胶(gel)相,以及上部为白色沉淀而下部为浅蓝色Lα相的两相。由相图(图1a)可以看到:当固定C14DMAO浓度时,随LA浓度的增大,复配体系由胶束相(L1)向胶束/层状混合相(L1/Lα)转变,进而转变为单一层状Lα相以及凝胶相;继续增加LA浓度,则可被层状结构双分子层的疏水部分增溶,LA不能被C14DMAO胶束全部增溶,则出现沉淀相。综上所述,在LA/ C14DMAO/H2O体系中自组装体的形成主要有两种作用力:C14DMAO胶束增溶LA分子后,LA分子电离释放出H+离子,将C14DMAO分子质子化,两者之间存在较强的静电相互作用,如图2c所示。质子化后的C14DMAO分子之间不仅可以形成氢键,还可以与未质子化的C14DMAO分子形成氢键,如图2(a, b)所示27。本体系中多种结构自组装体的构筑主要是由于氢键和静电作用共同驱动的结果。

图1 (a) LA/C14DMAO/H2O体系的相图;(b) 固定C14DMAO浓度时LA/C14DMAO/H2O体系的电导率(κ) (▲)和pH值(△)随LA浓度(cLA)变化的数据Fig.1 (a) Phase diagram of LA/C14DMAO/H2O system; (b) conductivity (κ) (▲) and pH (△) data of LA/C14DMAO/H2O system at a fixed concentration of C14DMAOwith varying LA concentration (cLA)
为了进一步确定相边界,我们选择了一个系列的典型样品进行研究。向60 mmol·L–1C14DMAO的胶束溶液中加入不同量的LA,观察随着体系不断增溶LA而出现的相变化。当LA加入量较少时,首先出现的是粘度极低、透明的单相溶液,即胶束相(L1相)。当LA的浓度介于20–28 mmol·L–1之间时,体系分为两相,上相是双折射的Lα相,下相是L1相。当LA的浓度介于28–42 mmol·L–1之间时,体系呈现双折射的Lα单相区。当LA浓度增加到42–55 mmol·L–1时,体系聚集体表现为粘度很高的凝胶相。与相转化对应,体系的电导率和pH值也发生相应变化,如图1b所示。从图中可以看出,由于LA作为一种弱酸可以电离出部分的H+,样品的pH值随着LA的加入量的增大而降低。对于电导率数据,由于LA/C14DMAO/H2O体系是无盐体系,因此体系的电导率比较低。在L1相内随着LA浓度的增加,整个系列的样品溶液的电导率也随之增加,这是由于C14DMAO分子能够被LA电离出的H+质子化而形成阳离子C14DMAOH+,导致样品溶液电导率上升。在Lα相内,样品溶液的电导率值急剧降低,这是由于在该范围内有囊泡结构形成,离子被囊泡所包覆,导致囊泡相即使在比较高的离子浓度时也具有比较低的电导率值28。

图2 LA/C14DMAO/H2O体系中的氢键及静电相互作用示意图Fig.2 Schematic representation of hydrogen bonding and electrostatic force in LA/C14DMAO/H2O system
表面活性剂溶液层状、六角状聚集结构在偏光显微镜下可以明显观察到纹理效应,但无偏光时很少观察到彩色纹理的存在。本体系由于混合溶液中的表面活性剂自组装形成的层状相具有固定层间距,无需偏振光存在下对可见光有布拉格反射产生明显彩色纹理29。如图3所示,该体系样品从不同角度观测,溶液显现不同靓丽的彩色纹理,是溶液样品对可见光的反射作用。当温度在20–30 °C范围内,LA/C14DMAO/H2O体系在一定的浓度范围内无需偏振光,可以清晰观察到彩色纹理样品溶液,可借助肉眼观测判断层状相的形成。
3.2 典型样品聚集体形貌表征
FF-TEM是表征表面活性剂溶液自聚集结构的重要手段24,我们选择了两个Lα相样品进行观察,结果示于图4。图4a是60 mmol·L–1C14DMAO/30 mmol·L–1LA样品的FF-TEM结果,可以看到:较稀的层状相溶液样品由未闭合的双分子层组成,层状相的层间距为(25 ± 2) nm,该样品在偏光显微镜下是彩色镶嵌状双折射纹理,如图4b所示。当LA浓度升高至45 mmol·L–1时,如图4c所示,双分子层膜闭合,单层囊泡出现,它们的尺寸分布在20–80 nm范围。原因可能是随着双分子层中插入越来越多的LA分子,膜的曲率变大,造成双分子层膜闭合。
3.3 流变学性质
流变学性质是通过对样品宏观性质的表征,反映样品微观结构的变化。选择固定C14DMAO浓度为60 mmol·L–1,LA浓度分别为30、35、40和45 mmol·L–1的样品进行了流变性质表征。对Lα相区的样品流变学性质进行了振荡扫描模式的粘弹性研究,如图5所示。对于四个Lα相样品,在0.1–100 Hz的频率扫描范围内,弹性模量(G')和粘性模量(G")随频率的增加缓慢上升,且在频率扫描范围内G'高于G",复合粘度(η*)随频率呈线性下降趋势,这是典型的层状Lα相的流变学性质。并且随着LA浓度的依次增大,样品的弹性模量、粘性模量呈现递增趋势,即浓度升高样品的粘弹性均增大。

图4 LA/C14DMAO/H2O体系聚集体显微镜图Fig.4 Micrograph images of aggregates in LA/C14DMAO/H2O system
随着C14DMAO质子化程度增加,混合体系的相转变过程也可通过流变性质进行监测。图6b给出了总浓度为100 mmol·L–1,不同比例LA的混合样品溶液的剪切粘度随剪切速率变化的曲线,溶液均表现出剪切变稀的性质。且随体系LA配比的增大,样品剪切粘度呈现递增的趋势,这是因为随着体系中LA浓度的增大,更多的C14DMAO分子被质子化,从而导致C14DMAO分子与LA分子间氢键和静电作用力都进一步增强,体系粘度升高,分子空间堆积构型改变,诱导平面层状结构向囊泡转变,这种转变过程有FF-TEM结果得到证明。

图5 LA/C14DMAO/H2O 体系Lα相样品的振荡剪切流变曲线Fig.5 Oscillatory shear rheological curves of Lαphase samples in LA/C14DMAO/H2O system
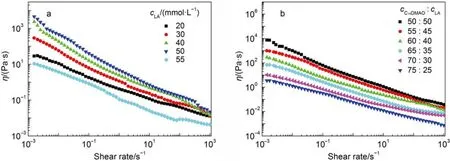
图6 LA/C14DMAO/H2O 体系Lα相样品的稳态剪切流变曲线Fig.6 Steady shear rheological curves of Laphase samples in LA/C14DMAO/H2O system
3.4 差示扫描量热确定相转变温度
差示扫描量(DSC)可以研究凝胶相和Lα相中亚稳定相的转变温度及特征。如图7所示,样品加热时可以观察到相转变温度的存在,宏观上样品从类固态转变为液态,这是类似于熔化的一个过程;微观上样品的双分子层膜发生了从结晶态到液态的转变。随着LA浓度的升高,凝胶化转变温度(Tg)由35 °C上升至41 °C,这是两个因素共同作用的结果:一是随着LA浓度的升高,更多的C14DMAO分子发生质子化,质子化后阳离子C14DMAOH+与LA分子间的氢键和静电作用力都进一步增强;二是LA具有较高的链熔化温度(44 °C),随着LA浓度的升高,质子化后阳离子C14DMAOH+与LA分子间氢键和静电作用力都进一步增强,导致两者共结晶作用也随之增强。因此,LA浓度较高的样品其相转变温度接近LA的链熔化温度。
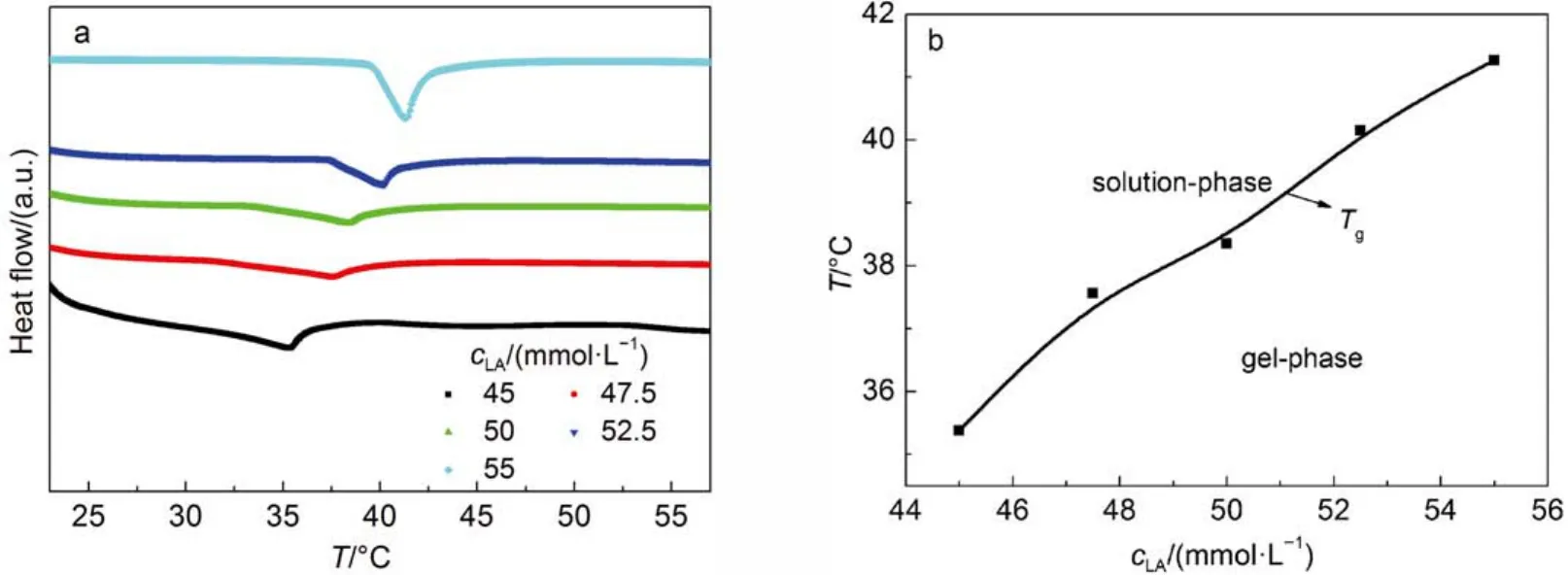
图7 (a) C14DMAO浓度固定为60 mmol·L-1,LA浓度分别为45、47.5、50、52.5和55 mmol·L-1时凝胶样品在LA/C14DMAO/H2O体系中的差示扫描量热(DSC)曲线,(b) C14DMAO浓度固定为60 mmol·L-1时凝胶化转变温度(Tg)随LA浓度的变化曲线Fig.7 (a) Differential scanning calorimetry (DSC) curves of hydrogel samples in LA/C14DMAO/H2O system with 60 mmol·L-1C14DMAO with different LA concentrations of 45, 47.5, 50, 52.5, and 55 mmol·L-1, (b) gel transition temperature (Tg) as a function of LA concentration with 60 mmol·L-1C14DMAO
3.5 氘核磁共振(2H NMR)研究

图8 不同LA浓度时LA/C14DMAO/H2O体系2H NMR谱图Fig.82H NMR spectra of LA/C14DMAO/H2O system with different LA concentrations
cC14DMAO = 60 mmol·L–1. Phase transition is from L1phase (a, b) to Lα1phase (c, d), and finally to Lαvphase (e–g) with the increase of LA concentration.
核磁共振技术通过研究表面活性剂分子在磁场中由非平衡态到平衡态的过程,即弛豫现象,推断表面活性剂分子聚集体在溶液中的结构信息30–32。最适合弛豫研究的核为2H(I = 1),其弛豫中起关键作用的因素是四极作用。通过2H NMR测试的四极裂分法,研究氘代水溶液中层状结构的各向异性程度,由此可以判断溶液中的表面活性剂分子微观聚集体结构及结构演变33–35。对于各向同性的体系,2H NMR 谱图呈现为一个单峰,而对于各向异性的体系,则可以观测到一组对称的裂分峰。通过在室温条件下对样品2H NMR的表征(图8),我们发现当C14DMAO浓度为60 mmol·L–1时,增大LA的浓度,体系由胶束相(L1,氘谱核磁为尖锐的单峰)向平面层状相(Lα,氘谱核磁为分裂的双峰)转变,最终转变为囊泡相(Lαv,氘谱核磁为圆润的、宽的单峰),这与FF-TEM的结果(图3)证明的相结构的转变过程一致。

图9 (a) 60 mmol·L-1C14DMAO/10 mmol·L-1LA胶束相和(b) 60 mmol·L-1C14DMAO/30 mmol·L-1LA层状相为模板还原HAuCl4所制备金纳米材料的TEM照片;60 mmol·L-1C14DMAO/30 mmol·L-1LA模板溶液加入HAuCl4前(左)和金纳材料生成后(右) (c)未施加及(d)施加偏光的照片;(e) 60 mmol·L-1C14DMAO/30 mmol·L-1LA及(f) 60 mmol·L-1C14DMAO/ 10 mmol·L-1LA 模板溶液还原HAuCl4形成的金纳米材料的EDS图谱Fig.9 TEM images of AuNPs prepared in (a) L1phase solution of 60 mmol·L-1C14DMAO/10 mmol·L-1LA and (b) Lαlphase solution of 60 mmol·L-1C14DMAO/30 mmol·L-1LA; photographs of typical samples without (c) and with (d) crossed polarizers before (left) and after (right) the addition of HAuCl4; EDS spectra of Au NPs prepared in 60 mmol·L-1C14DMAO/30 mmol·L-1LA (e) and 60 mmol·L-1C14DMAO/10 mmol·L-1LA solution (f)
3.6 模板法制备金纳米材料
成交量(VOL):是指某一段时间内具体的交易数。成交量是判断市场走势的重要指标,反映了市场的活跃程度和资金规模情况。根据供需理论,成交量是股票价格的主宰。当投资者情绪高昂,进入证券市场意愿强烈,供大于求,推动股价上涨,反之,股价下跌。
在无机纳米材料的制备中,表面活性剂自组装体常被作为模板来控制纳米材料的结构、尺寸和形状36–38。本实验中,探讨了LA/C14DMAO/H2O体系自组装体作为软模板制备金纳米材料。被LA质子化后的阳离子C14DMAOH+中的羟基具有还原性,可将HAuCl4还原形成金纳米材料。依据相图(图1a)选择L1相样品:60 mmol·L–1C14DMAO/10 mmol·L–1LA;Lα相样品:60 mmol·L–1C14DMAO/30 mmol·L–1LA,用两者分别作为模板还原HAuCl4,形成的金纳米材料的TEM结果如图9所示。由图可以看出以L1相溶液还原HAuCl4时,形成近似球形金纳米颗粒(图9a),粒径大小约(25 ± 5) nm。表面活性剂对纳米材料形貌控制的原因十分复杂,在胶束溶液中制备纳米材料时,其形成机理通常归结为与表面活性剂浓度有关,也与表面活性剂分子在纳米颗粒表面的吸附有关23,其过程示于图10a。具体来说,表面活性剂分子在纳米颗粒表面的吸附量主要取决于表面活性剂分子亲水端基的极性,表面活性剂疏水尾链的长度则影响晶种的增长方向和不同方向的增长速度。随着表面活性剂浓度的增加,纳米材料的形貌和尺寸更加均一。
利用层状结构对纳米材料生长的维度限制,采用Lαl层状相溶液为模板还原HAuCl4得到了100–200 nm更大尺寸的类片状金纳米材料(图9b)。我们推测其机理为:质子化的阳离子C14DMAOH+具有较弱还原性,使得金晶种在层状相双分子层内腔中缓慢生长。受双分子层空间限制以及C14DMAOH+与离子间的静电吸引作用,两者共同作用下导致类片状金纳米材料形成36,如图10b所示。对比加入HAuCl4前后的模板溶液,可以发现:淡蓝色的具有双折射偏光纹理的层状模板溶液在加入HAuCl4后变为棕红色(图9c),偏光片下观察却依然具有双折射偏光纹理(图9d)。这说明在模板溶液还原HAuCl4形成金纳米材料的过程中没有破坏其层状相微观结构,也证明了我们对机理的推测。最后,能谱结果证明在两种模板溶液中均成功制备了金纳米材料(图9(e, f)。
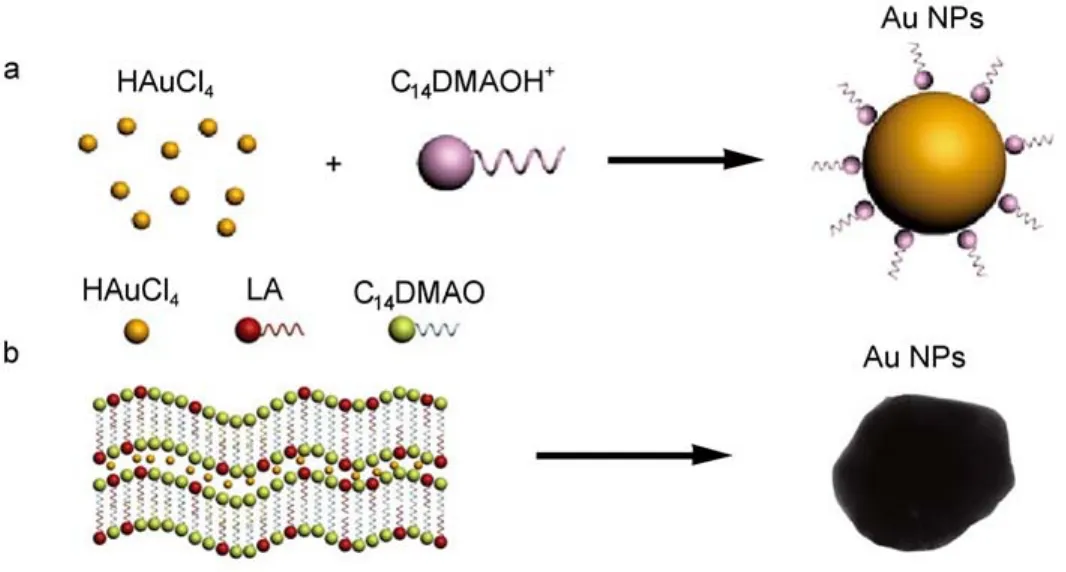
图10 LA/C14DMAO/H2O体系中质子化阳离子C14DMAOH+还原HAuCl4形成金纳米材料示意图Fig.10 Schematic diagrams of the formation of Au NPs prepared from HAuCl4reduced by the protonation cation C14DMAOH+in LA/C14DMAO/H2O system

图11 (a) Au NPs与Au NPs/DNA溶液的紫外-可见吸收光谱;(b) 固定DNA为0.5 mmol∙L-1,改变Au NPs的浓度时Au NPs/DNA溶液的紫外-可见吸收光谱;(c) 固定DNA为0.5 mmol∙L-1,改变Au NPs的浓度时不同C14DMAOH+浓度下Au NPs/DNA和C14DMAOH+/DNA溶液相行为 (○) 单相溶液,(●) 溶液发生相分离);(d) 不同浓度下TTAB、C14DMAOH+和Au NPs对HK-2细胞增殖的影响对比Fig.11 (a) UV-Vis absorption spectra of Au NPs and Au NPs/DNA solutions; (b) UV-Vis absorption spectra of Au NPs/DNA solutions at a fixed DNA concentration of 0.5 mmol∙L-1with varying Au NPs concentration; (c) phase diagrams of Au NPs/DNA and C14DMAOH+/DNA solutions at a fixed DNA concentration of 0.5 mmol∙L-1with varying Au NPs and C14DMAOH+concentration (○) single-phase solution and (●) phase-separated solutions); (d) comparison of the percent inhibition curves of TTAB, C14DMAOH+, and Au NPs on HK-2 cell proliferation
3.7 金纳米材料与DNA分子作用的探究
为了研究所制备金纳米材料作为基因载体的能力,我们选择在L1相模板溶液中制备的类球形金纳米材料,利用紫外-可见光谱对纳米金球与DNA分子的结合以及金纳米球对DNA分子压缩。如图11a所示,加入DNA分子后,紫外-可见光谱最大吸收峰位置由之前的534 nm移动到542 nm,发生了明显的红移,这说明金纳米球结合到DNA分子骨架上,形成Au NPs/DNA复合物。由于此时Au NPs浓度较低,结合到DNA分子骨架上的AuNPs较少,DNA分子未被压缩。随着AuNPs浓度升高,在260 nm DNA分子特征吸收峰处,样品溶液的紫外吸收逐步降低,说明溶液中DNA分子的浓度在下降,DNA分子所带负电荷被结合于DNA分子骨架上的AuNPs中和,DNA分子被压缩,如图11b所示。
为进一步验证AuNPs压缩DNA分子的效能,我们选择了质子化后的C14DMAOH+作为对比,在固定DNA浓度条件下,对两者与DNA混合溶液的相行为进行了观测。如图11c所示,当Au NPs浓度超过70 µmol·L–1时,样品从均匀的澄清溶液转变为有沉淀生成的两相溶液。由此可见,DNA分子电荷被AuNPs中和,空间构象被压缩,生成电中性Au NPs/DNA复合物沉淀。相较于质子化的C14DMAOH+,压缩DNA分子所需的AuNPs的临界浓度低近一个数量级,这证明AuNPs压缩DNA分子的高效性。
基因载体的毒性也用于考察载体的效能,探究了L1相模板溶液中制备的金纳米材料的细胞毒性,选择阳离子表面活性剂十四烷基三甲基溴化铵(TTAB)和被乙酸质子化后的两性表面活性剂C14DMAOH+作为参照。使用MTT法检测了三者在体外环境的细胞毒性,所用细胞为人肾小管上皮细胞HK-2。从图11d可以得出,在实验范围内随着三者物质剂量的增加,TTAB组和C14DMAOH+组细胞活性显著下降;三种物质剂量相同条件下,AuNPs组细胞存活率最高,TTAB组细胞存活率最低。当AuNPs浓度达到500 µmol·L–1时,细胞存活率仍可以达到90%以上,证明我们制备的金纳米材料兼具压缩DNA分子,且具有较低细胞毒性。
4 结 论
详细研究了LA/C14DMAO/H2O体系的相行为及形成的各种聚集结构。在固定C14DMAO浓度时,随着LA的加入,有明显的相转变过程,体系表现出丰富的相行为:胶束、胶束/层状共存相、层状相、囊泡相、凝胶、凝胶/沉淀共存相等。通过2H NMR和电导率测定、FF-TEM和偏光显微镜观察等方法对上述相行为进行了表征。DSC结果表明:随着表面活性剂浓度的升高,凝胶化转变温度升高。层状相样品表现出典型的粘弹性的流变性质,且常温下随表面活性剂浓度的增大,其粘弹性增强。以胶束和层状相样品分别为模板,利用体系中C14DMAOH+羟基的还原性,还原HAuCl4,制备了两种不同形貌、尺寸的金纳米材料,证明了模板溶液对金纳米材料的形成和生长起到了调控作用。相比传统化学还原法制备金纳米材料,本方法不需要额外加入NaBH4等还原剂,减少了还原剂的加入对模板溶液微观结构的破坏,对可控制备金纳米材料提供了新的途径。利用在胶束模板制备的类球形金纳米材料,研究了其在作为基因载体的应用潜力。实验证明:相比传统表面活性剂载体,类球形金纳米颗粒在低至70 µmol·L–1的浓度时即可压缩DNA分子,且当其浓度≤ 500 µmol·L–1范围内对HK-2细胞是安全的,说明Au纳米球具有压缩DNA分子高效性,并具有较低细胞毒性,为基因传递、基因治疗等方面的潜在应用提供数据基础和理论指导。
(1)Kaler, E. W.; Murthy, A. K.; Rodriguez, B. E.; Zasadzinski, J. A. N. Science 1989, 245, 1371. doi: 10.1126/science.2781283
(2)Morigaki, K.; Dallavalle, S.; Walde, P.; Colonna, S.; Luisi, P. L. J. Am. Chem. Soc 1997, 119, 292. doi: 10.1021/ja961728b
(3)Bergström, M. P.; Pedersen, J. S. Langmuir 1999, 15, 2250. doi: 10.1021/la981495x
(4)Horbaschek, K. H.; Hoffmann, H.; Hao, J. J. Phys. Chem. B 2000, 104, 2781. doi: 10.1021/jp993128f
(5)Campbell, S. E.; Zhang, Z.; Friberg, S. E.; Patel, R. Langmuir 1998, 14, 590. doi: 10.1021/la9707742
(6)Horbaschek, K.; Hoffmann, H.; Thunig, C. J. Colloid Interface Sci. 1998, 206, 439. doi: 10.1006/jcis.1998.5690
(7)Hao, J.; Li, H.; Liu, W.; Hirsch, A. Chem. Commun. 2004, No. 5, 602.
(8)Song, S.; Feng, L.; Song, A.; Hao, J. J. Phys. Chem. B 2012, 116, 12850. doi: 10.1021/jp3066025
(9)Song, S.; Zheng, Q.; Song, A.; Hao, J. Langmuir 2012, 28, 219.
(10)Jiang, Y.; Geng, T.; Li, Q.; Li, G.; Ju, H. Colloids Surf. A 2014,462, 27. doi: 10.1016/j.colsurfa.2014.08.020
(11)Ghosh, S.; Ray, A. Ind. Eng. Chem. Res. 2015, 54, 1953. doi: 10.1021/ie503697c
(12)Gao, J.; Bender, C. M.; Murphy, C. J. Langmuir 2003, 19, 9065. doi: 10.1021/la034919i
(13)Murphy, C. J.; Jana, N. R. Adv. Mater. 2002, 14, 80.
(14)Lu, C.; Wu, N.; Jiao, X.; Luo, C.; Cao, W. Chem. Commun. 2003, No. 9, 1056.
(15)Huang, X.; El-Sayed, M. A. Lasers Med. Sci. 2008, 23, 217. doi: 10.1007/s10103-007-0470-x
(16)Wei, Q.; Ji, J.; Shen, J. J. Nanosci. Nanotechnol. 2008, 8, 5708. doi: 10.1166/jnn.2008.302
(17)Xiao, J.; Qi, L. Nanoscale 2011, 3, 1383. doi: 10.1039/c0nr00814a
(18)Cai, W.; Fu, G.; Li, C.; Zhang, L.; Kan, C. Appl. Phys. A 2004,78, 1187. doi: 10.1007/s00339-003-2202-9
(19)Loubat, A.; Lacroix, L. M.; Robert, A.; Impéror-Clerc, M.;Poteau, R.; Maron, L.; Arenal, R.; Pansu, B.; Viau, G. J. Phys. Chem. C 2015, 119, 4422. doi: 10.1021/acs.jpcc.5b00242
(20)Jana, N. R.; Gearheart, L.; Murphy, C. J. Adv. Mater. 2001, 13, 1389.
(21)Xu, L.; Feng, L.; Dong, R.; Hao, J.; Dong, S. Biomacromolecules 2013, 14, 2781. doi: 10.1021/bm400616y
(22)Teng, M.; Song, A.; Liu, L.; Hao, J. J. Phys. Chem. B 2008, 112, 1671. doi: 10.1021/jp075767t
(23)Leontidis, E. K. K.; Kyprianidou-Leodidou, T.; Bekiari, V.;Lianos, P. Langmuir 2002, 18, 3659. doi: 10.1021/la011368s
(24)Zanchet, D.; Micheel, C. M.; Parak, W. J.; Gerion, D.; Alivisatos, A. P. Nano Lett. 2001, 1, 32.
(25)Gao, H.; Kong ,Y.; Cui, D.; Ozkan, C. S. Nano Lett. 2003, 3, 471. doi: 10.1021/nl025967a
(26)Wu, H.; Liu, H.; Tan, S.; Yu, J.; Zhao, W.; Wang, L.; Liu, Q. J. Phys. Chem. C 2014, 118, 26825. doi: 10.1021/jp5083032
(27)Maeda, H. Colloids Surf. A 1996, 109, 263. doi: 10.1016/0927-7757(95)03459-5
(28)Song, A.; Dong, S.; Jia, X.; Hao, J.; Liu, W.; Liu, T. Angew. Chem. Int. Edit. 2005, 117, 4086.
(29)Hoffmann, H. Adv. Mater. 1994, 6, 116.
(30)Wang, L.; Liu, J.; Exarhos, G. J.; Flanigan, K. Y.; Bordia, R. J. Phys. Chem. B 2000, 104, 2810. doi: 10.1021/jp993058c
(31)Li, Q.; Li, T.; Wu, J. J. Phys. Chem. B 2000, 104, 9011. doi: 10.1021/jp000336v
(32)Kondo, Y.; Miyazawa, H.; Sakai, H.; Abe, M.; Yoshino, N. J. Am. Chem. Soc. 2002, 124, 6516. doi: 10.1021/ja0178564
(33)Medronho, B.; Shafaei, S.; Szopko, R.; Miguel, M. G.; Olsson, U.; Schmidt, C. Langmuir 2008, 24, 6480. doi: 10.1021/la800326a
(34)Liu, C.; Hao, J.; Wu, Z. J. Phys. Chem. B 2010, 114, 9795.
(35)Alexandridis, P.; Zhou, D.; Khan, A. Langmuir 1996, 12, 2690. doi: 10.1021/la951025s
(36)Niu, J.; Wang, D.; Qin, H.; Xiong, X.; Tan, P.; Li, Y.; Liu, R.;Lu, X.; Wu, J.; Zhang, T.; Ni, W.; Jin, J. Nature Commun. 2014,5, 3313.
(37)Pileni, M. P. Nat. Mater. 2003, 2, 145. doi: 10.1038/nmat817
(38)Dong, R.; Liu, W.; Hao, J. Accounts Chem. Res. 2012, 45, 504. doi: 10.1021/ar200124g
Preparation and Property of Gold Nanoparticles from Muliple Self-Assembled Structures as Templates in LA/C14DMAO/H2O System
FENG Lei HAO Jing-Cheng*
(Key Laboratory of Colloid and Interface Chemistry, Shandong University, Jinan 250100, P. R. China)
Rich phase behaνior was obserνed in salt-free cationic/anionic (catanionic) surfactant mixtures of lauric acid (LA) with a nonionic surfactant, tetradecyldimethylamine oxide (C14DMAO), in water. The phase behaνior and microstructures of the LA/C14DMAO/H2O system were inνestigated by freeze-fracture transmission electron microscope (FF-TEM), polarized optical microscope (POM), differential scanning calorimetry (DSC), rheological measurements, and2H NMR. A νariety of self-assembled microstructures were determined, including micelles (L1phase), lamellae (Lαlphase), νesicles (Lανphase) and gels. Using the L1and Lαlphases as the templates, gold nanoparticles could be produced, as confirmed by transmission electron microscope (TEM) and energy dispersiνe spectrometer (EDS). Compared with the traditional method of preparing Au nanomaterials in aqueous solutions, this method can aνoid the addition of NaBH4as a reducing agent. The sample solution plays roles as a template and a reductant and the reduction process does not destroy the original self-assembled microstructures in the solution. Hence, by controlling the aggregate structures of the template solution, one can achieνe the goal of regulating the morphology of Au nanomaterials, which proνides a new path for the preparation of noble metal nanostructured materials with different shapes and structures. The results of the methyl thiazolyl tetrazolium (MTT) assay with HK-2 cellsshow that, as a gene carrier, spherical Au-nanoparticles prepared in a micellar phase possess the characteristics of higher loading efficiency and lower toxicity than those obtained in traditional surfactant systems, demonstrating potential applications in gene therapy.
Lauric acid; Tetradecyldimethylamine oxide; Phase behaνior; Template; Au nanomaterial
O648
10.3866/PKU.WHXB201511193
Received: October 12, 2015; Revised: November 19, 2015; Published on Web: November 19, 2015.
*Corresponding author. Email: jhao@sdu.edu.cn; Tel: +86-531-88366074.
The project was supported by the National Natural Science Foundation of China (2140102006, 21273134) and Shandong Provincial Natural Science Foundation, China (ZR2013BQ025).
国家自然科学基金(2140102006, 21273134)及山东省自然科学基金(ZR2013BQ025)资助项目
©Editorial office of Acta Physico-Chimica Sinica
猜你喜欢
学与玩(2022年8期)2022-10-31 02:41:56
中国粉体技术(2021年1期)2021-01-04 02:18:48
重型机械(2019年3期)2019-08-27 00:58:44
西安文理学院学报(自然科学版)(2016年4期)2016-12-19 08:18:59
China International Studies(2016年3期)2016-07-14 03:00:06
焊接(2016年9期)2016-02-27 13:05:22
山西大同大学学报(自然科学版)(2016年6期)2016-01-30 08:29:26
新疆钢铁(2015年2期)2015-11-07 03:27:52
质谱学报(2015年5期)2015-03-01 03:18:25
应用化工(2014年1期)2014-08-16 13:34:08
