
Castleman病合并系统性红斑狼疮的临床特征分析
2016-11-18 06:49:23曹欣欣王书杰周道斌
中国医学科学院学报 2016年5期
张 路,曹欣欣,王书杰,周道斌,李 剑
中国医学科学院 北京协和医学院 北京协和医院血液科,北京 100730
·论 著·
Castleman病合并系统性红斑狼疮的临床特征分析
张 路,曹欣欣,王书杰,周道斌,李 剑
中国医学科学院 北京协和医学院 北京协和医院血液科,北京 100730
目的 总结Castleman病(CD)合并系统性红斑狼疮(SLE)患者的临床特点。方法 根据北京协和医院病案室1994年至2014年整理的诊断信息,收集诊断符合CD病及SLE患者。以“Castleman’s disease”、“Systemic lupus erythematosus”、“SLE”、“lupus”为关键词在PubMed上检索1994至2014年间文献,筛选有详细临床资料记录的文献,临床资料不完善的病例被排除在本研究之外。结果 共纳入9例患者,其中北京协和医院2例,占该院同期确诊SLE患者的0.03%(2/6502),同期确诊CD患者的1.0%(2/100)。9例患者中,男2例,女7例,确诊CD时中位年龄39.0岁(21~60岁)。所有患者均为多中心型CD,病理检查提示浆细胞型3例,透明血管型3例,混合型3例。患者最常见症状为发热(88.9%,8/9),最常累及的系统为血液系统(88.9%,8/9),最常累及的脏器为肾脏(88.9%,8/9)。55.6%(5/9)的患者存在自身免疫性血小板减少,显著高于普通SLE患者的15.0%(P<0.01)。所有患者均未出现中枢神经系统受累证据。结论 CD合并SLE是一种较为罕见的临床情况。与普通SLE患者相比,合并CD的SLE患者可能更易出现血小板减少,较少出现神经系统受累。
Castleman病;系统性红斑狼疮;自身免疫性血小板减少
ActaAcadMedSin,2016,38(5):543-547
Castleman病(Castleman’s disease,CD)是一种较为少见的淋巴增生性疾病[1]。临床上根据肿大淋巴结分布和器官受累情况不同,可分为单中心型(unicentric CD,UCD)和多中心型(multicentric CD,MCD)。前者常仅累及单个淋巴结区域,相关症状较轻,外科治疗效果良好;后者则累及多个淋巴结区域,有较为明显的系统性症状。CD合并系统性红斑狼疮(systemic lupus erythematosus,SLE)虽是一种较为罕见的临床情况,但越来越多的学者认为诸如SLE的系统性炎症性疾病可能是CD的重要病因[2]。因此,描述合并SLE的CD患者临床特征,有助于加深对于CD这一少见疾病的认识。本研究分析了近20年来北京协和医院及文献报道的所有此类病例,总结了CD合并SLE患者的临床特点,以期为此类患者的临床诊疗提供帮助。
资料和方法
资料来源 根据北京协和医院病案室1994年至2014年整理的诊断信息,收集诊断符合CD及SLE患者共2例。以“Castleman’s disease”、“systemic lupus erythematosus”、“SLE”、“lupus”为关键词在PubMed上检索1994至2014年间文献,筛选有详细临床资料记录的文献,临床资料不完善的病例被排除在本研究之外。收集入选患者的临床特点(包括性别、年龄、症状、系统累及情况、实验室检查、病理结果、治疗方法)及转归信息(好转或死亡),总结患者的临床特点,并与一般SLE患者进行比较。
诊断标准 SLE的诊断采用1982年美国风湿病学会分类标准[3];CD的诊断依赖病理,局限型疾病(仅有单处淋巴结肿大)诊断为UCD,病变累及2个或以上的淋巴结/区域则诊断为MCD[4]。
统计学处理 采用SPSS 13.0统计软件,连续变量以中位数(范围)表示,两分变量采用百分率表示,患者临床特点与一般SLE患者比较采用二项分布检验,P<0.05为差异有统计学意义。
结 果
一般情况 共纳入9例患者,其中,7例来自文献[5- 10];2例来自北京协和医院,占该院同期确诊SLE患者的0.03%(2/6502),同期确诊CD患者的1.0%(2/100)。9例患者中,男2例,女7例,中位年龄39.0岁(21~60岁)(表1)。
临床特征 所有患者均有多发外周淋巴结肿大,符合MCD。最常见的症状为发热(88.9%,8/9),最常累及的系统为血液系统(88.9%,8/9),最常累及的脏器为肾脏(88.9%,8/9)。55.6%(5/9)的患者患关节炎。自身免疫性血小板减少(autoimmune thrombocytopenia,AITP)较为常见,有55.6%(5/9)的患者出现了血小板降低。所有患者均未出现中枢神经系统受累(表1)。
病理检查 病理检查结果提示,9例患者中,3例为浆细胞型,3例为透明血管型,3例为混合型(表1)。
实验室检查 所有患者的抗核抗体(antinuclear antibody,ANA)均阳性,66.7%(6/9)的患者抗-SSA抗体(anti-SSA)抗体阳性,66.7%(6/9)的患者抗双链DNA阳性,44.4%(4/9)的患者Coombs试验阳性,33.3%(3/9)的患者抗心磷脂抗体(anticardiolipin antibody,ACL)阳性(表1)。
治疗及转归 66.7%(6/9)的患者单用糖皮质激素治疗,33.3%(6/9)的患者使用了以CHOP方案(环磷酰胺+多柔比星+长春新碱+泼尼松,CHOP)或COP方案(环磷酰胺+长春新碱+泼尼松,COP)方案为基础的化疗,其中1例患者使用了利妥昔单抗治疗。8例患者有转归方面信息,除1例患者(60岁男性,有肾脏及血液系统受累,单用糖皮质激素)死亡外,其余7例预后良好(表1)。
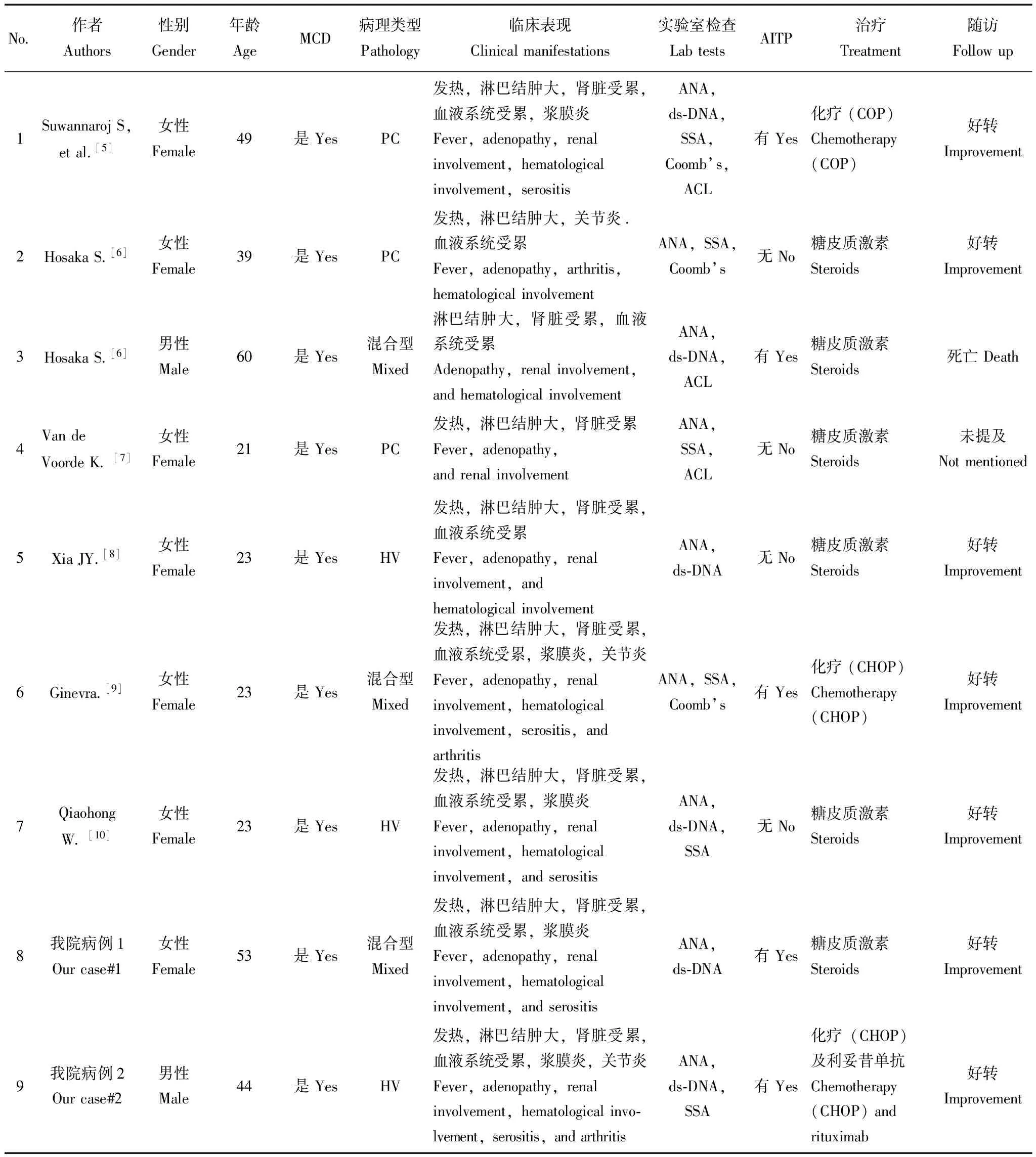
表 1 CD合并SLE病例汇总
CD:Castleman病;SLE:系统性红斑狼疮; MCD:多中心型Castleman病; AITP:自身免疫性血小板减少; PC:浆细胞型; HV:透明血管型; ANA:抗核抗体; ds-DNA:抗双链DNA抗体; SSA:抗SSA抗体; ACL:抗心磷脂抗体; COP:环磷酰胺+长春新碱+泼尼松/泼尼松龙; CHOP:COP+柔红霉素;肾脏受累:24 h尿蛋白>0.5 g或次尿蛋白≥3+,或有细胞管型; 血液系统受累:溶血性贫血或白细胞减少(<4000/μl)或淋巴细胞减少 (<1500/μl) 或血小板减少(<100 000/μl)(排除药物影响); 浆膜炎:超声心动或CT提示胸膜炎或心包炎(或浆膜腔积液)
CD:Castleman’s disease;SLE:systemic lupus erythematosus; MCD:multicentric CD; AITP:autoimmune thrombocytopenia; PC:plasma variant; HV:hyaline-vascular variant; ANA:antinuclear antibodies; ds-DNA:anti-double-strand DNA; SSA:anti-SSA antibody; ACL:anti-cardiolipid antibody; COP:cyclo-phosphamide+vincristine+prednisone; CHOP:COP+daunorubicin;renal involvement:24 h urine protein>0.5 g or urine protein≥3+,or with casts; hematological involvement:hemolytic anemia or leucopenia (<4000/μl) or lymphopenia (<1500/μl) or thrombocytopenia (<100 000/μl); serositis:pleuritis or pericarditis (or serous effusion) detected by cardiac ultrasound or CT
二项分布检验结果 采用二项分布将本组患者系统受累以及实验室检查结果与普通SLE患者(血液系统受累85%,肾脏受累50%,浆膜炎60%,关节炎60%,ANA阳性率98%)进行比较,结果显示,合并CD的患者在前述临床表现上,与普通SLE患者差异无统计学意义(P>0.05)。本组患者的AITP发生率为55.6%,明显高于普通SLE患者的15.0%[9](P<0.01)。
典型病例 病例1:女,53岁,因“淋巴结肿大7月余,发热1周”入院。查体:双侧颈部、腋窝淋巴结肿大,肝脾不大。实验室检查:血红蛋白(hemoglobin,Hb)99 g/L,血小板(platelet,PLT)90×109/L,血肌酐(serum creatinine,SCr)128 μmol/L,ANA/anti-dsDNA/Coombs试验(+),尿蛋白(+)(3.01 g/24 h)。右侧腋窝淋巴结病理:CD(混合型)。肾穿病理:狼疮肾炎IV型。诊断“CD(多中心型、混合型),SLE,狼疮肾炎IV型”。予泼尼松及环磷酰胺治疗,治疗后患者症状消失,血象恢复正常,ANA、抗dsDNA、Coombs转阴,随诊1.5年病情无复发。病例2:男,44岁,因“发热40 d,淋巴结和肝脾肿大30 d”入院。查体:双侧颈部、腋窝、腹股沟淋巴结,肝、脾肿大,腹水征(+)。实验室检查:PLT 9×109/L,SCr 180 μmol/L,ANA/抗dsDNA/抗SSA(+),尿蛋白(+)(0.68 g/24 h)。骨髓涂片:巨核细胞849个、无产板型,血小板少见。颈部淋巴结病理:CD(透明血管型)。诊断“CD(多中心型、透明血管型),SLE,AITP”。予CHOP方案治疗,因血小板回升不满意,后加用利妥昔单抗。治疗后患者症状消失,血小板恢复正常,ANA、抗dsDNA、抗SSA转阴,随诊7年病情无复发。
讨 论
CD合并SLE在1994年首次被报道[6],而之后陆续的此类病例报道[5- 10]证实该现象并非偶然。作为一种淋巴增生性疾病,除SLE外,尚有报道发现CD与包括干燥综合征、类风湿关节炎、混合性结缔组织病、重症肌无力等存在关联[7]。对于此种关联存在多种解释:(1)强调CD的重要性,认为CD可能与其他涉及B细胞的疾病(如多发性骨髓瘤、慢性淋巴细胞白血病等)类似,通过直接产生自身抗体导致自身免疫性疾病(如副肿瘤性天疱疮、自身免疫性溶血性贫血[12]等)或自身免疫性疾病相关表现(如ANA阳性);(2)认为包括SLE在内的系统性炎症性疾病可能是CD的重要病因[2],尤其是对于不伴HIV和人类疱疹病毒- 8(human herpes virus- 8,HHV- 8)的特发性MCD(idiopathic MCD,iMCD)患者。具体机制可能为:系统性炎症疾病引起自身免疫系统激活、细胞因子(如白细胞介素- 6、血管内皮生长因子等)释放,从而引起浆细胞增生、新生血管形成,导致CD发生。虽然存在机制上的可能相关性,但就北京协和医院收治患者情况来看,SLE患者中合并CD的比例极低(0.03%),低于既往文献中报道的比例,有文献报道伴体表淋巴结肿大的SLE患者,淋巴结活检病理提示26%的标本存在类似CD的病理改变[13]。上述差异可能与临床诊疗习惯有关:对于采用无创手段即可诊断的SLE患者,若伴有肿大的淋巴结,即使确实为CD,大部分患者也可通过针对SLE的治疗(单用糖皮质激素或加用免疫抑制剂)达到淋巴结缩小的效果;因此,对于确诊SLE且治疗有效的患者,往往不再进行有创性病理检查,故可能漏诊相当部分的CD患者。不过基于本研究文献复习的结果以及CD和SLE之间潜在的病因学关系,我们有理由推测:合并淋巴结肿大的SLE患者,可能有相当部分患者都存在CD。因此,我们建议对于淋巴结肿大的SLE患者,可在权衡利弊的情况下考虑淋巴结活检,一方面可以提高潜在CD的发现率;另一方面:考虑到随着CD认识的不断深入,新的针对性治疗方法逐渐涌现[14],对患者进行精确诊断,可指导更精准的治疗策略,从而改善患者预后。
本组资料显示,近20年来,北京协和医院住院治疗的200名CD患者中,合并SLE的发生率为1.0%。CD是一种罕见疾病,2011年前多为病例报道[15],2011年后才逐渐出现较大宗病例系列。即使目前最大宗的病例报道[12](114例),也只提到了6例患者合并自身免疫性溶血性贫血以及6例合并AITP,并无合并SLE发生率的数据。其他患者数较少的CD系列与此类似,也没有提及患者中SLE的发生率。Frizzera等[16]曾报道了15例CD患者,其中6例存在与SLE相关的症状,但均不满足4条或以上的SLE分类标准。
本研究发现合并CD的SLE患者,血小板减低的发生率更高。考虑到CD本身便会引起自身免疫性血小板减低[17],该现象似乎并不难解释。CD合并AITP的机制可能为与CD合并SLE的机制类似[9,17]。从本研究收集的9例患者来看,共有4例合并AITP,血小板减少程度多样,既有重度血小板减少(9×109/L),亦有轻-中度血小板减少,体现出疾病的异质性。
本研究还发现合并CD的SLE患者罕有中枢神经系统受累。这可能是合并CD的SLE患者的特点之一。但需要指出的是:(1)与血液系统、肾脏受累不同(有相对简单且客观的实验室指标),判断SLE患者是否存在中枢神经系统受累,筛查方法复杂且多样,依据筛选方法不同,结果差异较大[18];(2)本研究大多数病例来自已报道文献,对于较为繁复的神经系统表现难于详细收集,可能在判断是否存在中枢神经系统受累方面存在遗漏。因此,该现象(较少中枢神经系统受累)是否真实存在于合并CD的SLE患者中,尚需积累更多的患者进行观察。
综上,CD合并SLE是一种较为罕见的临床情况,与普通SLE患者相比,合并CD的SLE患者更易出现血小板减少,较少出现神经系统受累。考虑到本研究是一项病例数较少的回顾性研究,上述结论还有待后续研究进一步证实。
[1]Castleman B,Iverson L,Menendez VP. Localized mediastinal lymphnode hyperplasia resembling thymoma[J]. Cancer,1956,9(4):822- 830.
[2]Fajgenbaum DC,van Rhee F,Nabel CS. HHV- 8-negative,idiopathic multicentric Castleman disease:novel insights into biology,pathogenesis,and therapy[J]. Blood,2014,123(19):2924- 2933.
[3]Tan EM,Cohen AS,Fries JF,et al. The 1982 revised criteria for the classification of systemic lupus erythematosus[J]. Arthritis Rheum,1982,25(11):1271- 1277.
[4]Dispenzieri A, Armitage JO,Loe MJ,et al. The clinical spectrum of Castleman’s disease[J]. Am J Hematol,2012,87(11):997- 1002.
[5]Suwannaroj S,Elkins SL,McMurray RW. Systemic lupus erythematosus and Castleman’s disease[J]. J Rheumatol,1999,26(6):1400- 1403.
[6]Hosaka S,Ryumachi KH. Three cases of Castleman’s disease mimicking the features of collagen disease[J]. Ryumachi,1994,34(1):42- 47.
[7]Van de Voorde K,De Raeve H,De Block CE,et al. Atypical systemic lupus erythematosus or Castleman’s disease[J]. Acta Clin Belg,2004,59(3):161- 164.
[8]Xia JY,Chen XY,Xu F,et al. A case report of systemic lupus erythematosus combined with Castleman’s disease and literature review[J]. Rheumatol Int,2012,32(7):2189- 2193.
[9]De Marchi G,De Vita S,Fabris M,et al. Systemic connective tissue disease complicated by Castleman’s disease:report of a case and review of the literature[J]. Haematologica,2004,89(4):ECR03.
[10]Qiaohong W,Huaxiang W,Shengdong L. Systemic lupus erythematosus complicated by Castleman’s disease:a case report[J]. Chin J Intern Med,2008,47(3):244.
[11]Fauci AS,Braunwald E,Kasper DL,et al. Harrison’s internal medicine[M]. 17th ed. New York:McGraw-Hill,2007:2075- 2082.
[12]Dong Y,Wang M,Nong L,et al. Clinical and laboratory characterization of 114 cases of Castleman disease patients from a single centre:paraneoplastic pemphigus is an unfavourable prognostic factor[J]. Br J Haematol,2015,169(6):834- 842.
[13]Kojima M,Nakamura S,Morishita Y,et al. Reactive follicular hyperplasia in the lymph node lesions from systemic lupus erythematosus patients:A clinicopathological and immunohistological study of 21 cases[J]. Pathol Int,2000,50(4):304- 312.
[14]van Rhee F,Wong RS,Munshi N,et al. Siltuximab for multicentric Castleman’s disease:a randomised,double-blind,placebo-controlled trial[J]. Lancet Oncol,2014,15(9):966- 974.
[15]Talat N,Schulte KM. Castleman’s disease:systematic analysis of 416 patients from the literature[J]. Oncologist,2011,16(9):1316- 1324.
[16]Frizzera G,Peterson BA,Bayrd ED,et al. A systemic lymphoproliferative disorder with morphologic features of Castleman’s disease. Clinical findings and clinicopathological correlations in 15 patients[J]. J Clin Oncol 1983,3(9):1202- 1216.
[17]Sood R,Taylor HC,Daw H. Multicentric Castleman’s disease,associated with idiopathic thrombocytopenic purpura[J]. Case Rep Hematol,2013,2013:269268.
[18]Hanly JG. ACR classification criteria for systemic lupus erythematosus:limitations and revisions to neuropsychiatric variables[J]. Lupus,2004,13(11):861.
Clinical Features of Patients with Castleman’s Disease Complicated bySystemic Lupus Erythematosus
ZHANG Lu,CAO Xin-xin,WANG Shu-jie,ZHOU Dao-bin,LI Jian
Department of Hematology,PUMC Hospital,CAMS and PUMC,Beijing 100730,China
LI Jian Tel:010- 69155549,E-mail:lijian@pumch.cn
Objective To investigate the clinical features of patients with Castleman’s disease (CD) and systemic lupus erythematosus (SLE). Methods According to the diagnostic information between 1994 to 2014 extracted from the database of the Medical Record Department of Peking Union Medical College Hospital (PUMCH),patients with CD and SLE were included. A thorough literature review utilizing the key words of “Castleman’s disease”,“systemic lupus erythematosus”,“SLE”,and “lupus” was performed in PubMed during the same period. Cases with detailed clinical information were included while cases without detailed information were excluded from the analysis of this study. Results Nine patients worldwide were available for analysis [2 cases from PUMCH,accounted for 0.03%(2/6502) of all patients diagnosed as SLE and 1.0% (2/100) of patients diagnosed as CD during the same period] with a male-to-female ratio of2∶7. The median age at diagnosis of CD was 39.0 years (range:21- 60 years). All patients were diagnosed as multicentric CD with generalized peripheral lymphadenopathy. Pathologic examination showed a balanced distribution:plasma cell variant:hyaline-vascular variant:mixed variant=3∶3∶3. Fever was the most common symptom (88.9%,8/9). Blood system was the most commonly involved system (88.9%,8/9) and kidneys were the most commonly involved organ (88.9%,8/9). Autoimmune thrombocytopenia (AITP) was observed in 55.6% (5/9) of patients,which was significantly higher than the general SLE patients (15.0%) (P<0.01). None of the 9 patients had evidence of central nervous system involvement. Conclusions CD complicated by SLE is a rare clinical condition. Compared to the general SLE population,this subgroup of patients may have higher rate of AITP and lower rate of central nervous system involvement.
Castleman’s disease; systemic lupus erythematosus; autoimmune thrombocytopenia
北京协和医院杰出青年基金项目(JQ201508) Supported by the Peking Union Medical College Hospital Outstanding Youth Funding Project (JQ201508)
李 剑 电话:010- 69155549,电子邮件:lijian@pumch.cn
R559;R593.24+1
A
1000- 503X(2016)05- 0543- 05
10.3881/j.issn.1000- 503X.2016.05.009
2015- 08- 31)
猜你喜欢
中国毕业后医学教育(2021年1期)2021-12-06 06:46:28
中国民间疗法(2021年13期)2021-08-30 08:56:34
中国毕业后医学教育(2021年6期)2021-08-19 03:24:02
中国新闻周刊(2021年9期)2021-03-29 20:33:56
天津医科大学学报(2021年2期)2021-03-29 05:31:10
现代临床医学(2019年6期)2019-12-07 06:03:42
中国循证儿科杂志(2019年1期)2019-03-29 01:34:28
中国男科学杂志(2016年5期)2016-12-01 05:20:21
名作欣赏(2014年29期)2014-02-28 11:24:31
西南军医(2014年1期)2014-02-03 03:06:37
