
新型离子液体负载手性硫代咪唑啉酮的合成
2016-11-18 09:29梁现蕊陈鑫磊
浙江工业大学学报 2016年5期
梁现蕊,陈鑫磊
(1.浙江工业大学 绿色制药协同创新中心,浙江 杭州 310014;2.浙江工业大学 药学院,浙江 杭州 310014)
新型离子液体负载手性硫代咪唑啉酮的合成
梁现蕊1,2,陈鑫磊1,2
(1.浙江工业大学 绿色制药协同创新中心,浙江 杭州 310014;2.浙江工业大学 药学院,浙江 杭州 310014)
设计并合成了新型离子液体负载的手性硫代咪唑啉酮,以L-苯丙氨酸为手性原料,依次经过Boc保护、酰胺化反应、脱Boc反应和环化反应制备了(S)-5-苄基-2,2-二甲基-3-(1-咪唑基丙基)-4-氧咪唑烷酮,进一步经Lawesson试剂硫代,在甲苯溶剂中80 ℃下反应8 h,得到的手性硫代咪唑啉酮再与正丁基碘在乙腈溶液中回流反应24 h,首次制得了新型离子液体负载的手性硫代咪唑啉酮,并取得了较佳的立体选择性.其中,实验通过对脱Boc反应进行改进,即采用氯化氢的二氧六环溶液为反应试剂进行脱Boc反应,优化反应条件,获得了较佳的收率.
硫代咪唑啉酮,脱Boc,离子液体
手性(Chiral)是自然界的基本属性之一,其在药物化学中也具有重要意义,不同构型的手性异构体在药理活性上可能具有非常大的差异[1],因此,单一构型手性药物的合成,在手性药物研究中具有十分重要的意义.不对称合成是制备手性化合物的重要方法之一,通过手性催化剂的增值作用,可以大量地合成手性目标化合物.因此,不对称催化是当前有机合成和催化科学的前沿研究领域[2].
在过去的40多年里,不对称催化技术发展迅速,成为占有主导地位的不对称合成方法.不对称催化技术又包括酶催化、手性金属配合物催化以及有机小分子催化.手性咪唑啉酮催化剂是继脯氨酸之后又一手性胺类催化剂,自MacMillan合成并报道以来[3],由于其优良的催化特性引起了广泛的关注,成功用于不对称Diels-Alder反应[3-5]、不对称1,3-偶极环加成[6]、不对称Friedel-Crafts烷基化反应[7-8]、醛α位的不对称取代反应[9-11]和不对称ene反应[12]等多种反应中.在该催化剂研究的基础上,我们课题组对咪唑啉酮催化剂进行结构修饰,成功开发出了手性硫代咪唑啉酮.由于硫原子比氧原子半径大,硫代后,硫代酰胺中的电荷更易于从N转移到S,致使C-N键更稳定,因此推测硫代咪唑啉酮催化剂具备更好的催化活性.实验研究结果表明:该催化剂成功在吡咯、吲哚等的不对称Friedel-Crafts烷基化反应、醛的α氧化反应以及Diels-Alder等等反应中表现了良好的催化活性[13-16].近年来,对有机手性小分子结构的修饰仍然有较成功的例子,其中离子液体负载的手性咪唑啉酮催化剂取得了较好的应用[17].离子液体作为可溶性载体[18],在不对称合成和拆分方面都有应用[19-20],与其他常见的可溶性聚合物载体相比,其分子量较小;在二氯甲烷、四氢呋喃、甲醇等溶剂中具有良好的溶解性,但在正己烷、乙醚等溶剂中不溶且能快速析出
固体[21],由此,离子液体负载以后使催化剂更容易回收且可循环使用.受上述研究成果的启发,本实验开发了一种新型离子液体负载的手性硫代咪唑啉酮类催化剂.在已开发的手性硫代咪唑啉酮催化剂的基础上,对其结构进行修饰,即在其结构中增加一个有机离子基团,使其同时具有手性催化活性和离子液体的性质.
1 实验部分
1.1 催化剂设计
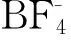

1.2 仪器与试剂
仪器:核磁共振氢谱在Varian 400(400 MHz)核磁共振仪上测定,CDCl3做溶剂,TMS为内标.ee值在Agilent 1100高效液相上测定.分子量均在Thermo Finnigan LCQ Advantage质谱仪上测定.
试剂:二碳酸二叔丁酯(Boc)2O、1-(3-二甲氨基丙基)-3-乙基碳二亚胺盐酸盐(EDC)、1-羟基苯并三唑(HOBt)、对甲苯磺酸(p-TSA)、Lawesson试剂(LR)等,如未特别提及均为市售分析纯,使用前均不用特别处理.
1.3 实验方法
1.3.1 N-(1-咪唑丙基)苯丙氨酰胺(Ⅳ)的合成
以L-苯丙氨酸为原料,按文献方法先对氨基酸的氨基进行Boc保护得到化合物Ⅱ,再进行酰胺化反应得到化合物Ⅲ.在100 mL单口圆底烧瓶中加入化合物Ⅲ(3.72 g,10 mmol),加入1 M氯化氢的二氧六环溶液40 mL,室温磁力搅拌24 h,反应过程中用薄板层析(TLC)跟踪检测,展开剂组成为:V(CH2C12)∶V(PE)∶V(EtOAc)∶V(MeOH)=1∶1∶1∶0.5,Rf=0.25.反应结束后,蒸干溶剂,加适量水,用饱和Na2CO3调节pH至10,用CH2Cl2萃取(30 mL×3),合并有机相并用Na2SO4干燥,过滤后蒸干溶剂,得黄色粘稠液体2.72 g,为化合物Ⅳ,产率99%,质量分数98%(HPLC面积归一法测得).其中1 M氯化氢的二氧六环溶液制备过程如下:准备两个两口烧瓶两口烧瓶,在其中一个中加入100 mL二氧六环,在HCl发生器的另一两口烧瓶中加入食盐,恒压滴液漏斗中加入浓硫酸缓慢逐滴滴加,生成的HCl气体通入二氧六环中,直至二氧六环烧瓶瓶重增加8.8 g.1H NMR(400 MHz, CDCl3)δ7.47(s,1H),7.40(s,1H),7.31~7.28(t,J=7.1 Hz,2H),7.26~7.22(d,J=7.1 Hz,2H),7.22~7.17(t,J=7.1 Hz,2H),7.03(s,1H),6.91(s,1H),3.94~3.90(t,J=6.9 Hz,2H),3.61~3.58(dd,J=8.9,4.2 Hz,1H),3.34~3.21(m,2H),3.20~3.19(d,J=4.2 Hz,1H),2.76~2.70(dd,J=13.6,8.9 Hz,1H),2.00~1.93(m,4H).MS(ESI,m/z):273[M+H]+.
1.3.2 (S)-5-苄基-2,2-二甲基-3-(咪唑丙基)-4-氧咪唑烷酮(Ⅴ)的合成
在250 mL圆底烧瓶(装有回流冷凝管,氮气保护装置)中一次性加入化合物Ⅳ(2.72 g,10 mmol),丙酮(70 mL),对甲苯磺酸(200 mg,1 mmol),氮气保护,80 ℃回流反应,反应过程中用薄板层析(TLC)跟踪检测,展开剂组成为:V(CH2C12)∶V(PE)∶V(EtOAc)∶V(MeOH)=1∶1∶1∶0.5,Rf=0.4,反应12 h后结束,蒸干溶剂,加入饱和NaHCO3溶液,使用CH2Cl2萃取(30 mL×3),合并有机相并用Na2SO4干燥,过滤,蒸干溶剂,产物用硅胶柱层析分离,展开剂组成为:V(CH2C12)∶V(PE)∶V(EtOAc)∶V(MeOH)=1∶1∶1∶0.2,得黄色粘稠液体2.47 g,为化合物V,产率79.2%.1H NMR(400 MHz,CDCl3)δ 7.47(s,1H),7.30~7.21(m, 5H),7.05(s,1H),6.91(s,1H),4.05~3.80(m,2H),3.79~3.77(t,J=5.2 Hz,1H),3.40~3.32(m,1H),3.16~3.01(m,2H),2.94~2.73(m,1H),2.12(s,1H),2.02~1.84(m,2H),1.23(s,3H),MS(ESI,m/z):313[M+H]+.
1.3.3 (S)-5-苄基-2,2-二甲基-3-(咪唑丙基)-4-硫咪唑烷酮(Ⅵ)的合成
在50 mL单口圆底烧瓶(装有回流冷凝管,氮气保护装置)中一次性加入化合物V(3.12 g,10 mmol),甲苯(60 mL),Lawesson试剂(2.4 g,6 mol),氮气保护,80 ℃反应,反应过程中用薄板层析(TLC)跟踪检测,展开剂组成为:V(CH2C12)∶V(PE)∶V(EtOAc)∶V(MeOH)=1∶1∶1∶0.5,Rf=0.5,24 h反应结束,蒸干溶剂,用硅胶柱层析分离,展开剂组成为:V(CH2C12)∶V(PE)∶V(EtOAc)∶V(MeOH)=1∶1∶1∶0.2得黄色粘稠液体0.73 g,为化合物Ⅵ,产率22%,ee值99%(HPLC∶OD-H,V(正己烷)∶V(异丙醇)=70∶30,v=0.8 mL/min,λ=210 nm).1H NMR(400 MHz,CDCl3)δ8.00(s,1H),7.38~7.25(m,5H),6.99(s,1H),6.85(s,1H),3.89~3.83(m,2H),3.82~3.80(t,J=1.9 Hz,1H),3.77~3.72(m,1H),3.70~3.64(m,1H),3.53~3.52(m,1H),2.31~2.21(m,2H),1.39(s,3H),1.35(s,3H).MS(ESI,m/z):327[M-H]-.
1.3.4 离子液体负载手性硫代咪唑啉酮(Ⅶ)的合成
在25 mL单口圆底烧瓶(装有回流冷凝管)中加入手性硫代咪唑啉酮Ⅵ(0.64 g,2 mmol),正丁基碘(0.40 g,2.2 mmol)乙腈(10 mL),80 ℃下回流反应,反应过程中用薄板层析(TLC)跟踪检测,展开剂组成为:V(CH2C12)∶V(PE)∶V(EtOAc)∶V(MeOH)=1∶1∶1∶0.5,Rf=0.1,24 h反应结束,蒸干溶剂,得黄色粘稠液体,为化合物Ⅶ,即碘化离子液体负载的手性硫代咪唑啉酮,产率75%.1H NMR(400 MHz,CDCl3)δ9.94(s,1H),9.79(s,1H),9.41(s,1H),7.48~6.99(m,5H),4.79~4.64(m,3H),4.13~4.03(m,2H),3.84~3.74(m,1H),3.70~3.60(m,1H),3.08~3.00(m,1H),2.98~2.84(m,4H),2.39~2.30(m,1H),2.23~2.15(s,6H),1.67~1.53(m,2H),1.27~1.23(t,J=7.1Hz,3H).MS(ESI,m/z):381[M-I]+.
上述得到的产物分别与KBF4和KPF6进行阴离子置换反应,液体负载的手性硫代咪唑啉酮,产率达99%.在50 mL单口圆底烧瓶中加入上述碘化离子液体负载的手性硫代咪唑啉酮(0.98 g, 2 mmol),KBF4(0.38 g, 3 mmol),H2O(10 mL),丙酮(10 mL),室温下磁力搅拌过夜.反应结束后蒸干丙酮,水相用CH2Cl2萃取(15 mL×3)萃取,合并有机相并用水洗3次,用Na2SO4干燥,过滤,蒸干溶剂得黄色粘稠液体,为四氟硼酸化离子液体负载的手性硫代咪唑啉酮,产率99%.其六氟磷酸化离子液体负载的手性硫代咪唑啉酮也是用相同方法制得,为棕黄色粘稠液体.
同时,采用与正丁基碘反应同样的方法,将正丁基碘换成正丁基氯,即可得氯化离子液体负载的手性硫代咪唑啉酮.
2 结果讨论
本研究以L-苯丙氨酸为起始原料,经过Boc保护、酰胺化反应、脱Boc反应、环化反应、硫代和离子化6步反应制得离子液体负载手性硫代咪唑啉酮VII催化剂.该反应路线整体转化率高,其中Boc保护反应可以定量转化,收率达99%以上;影响酰胺化反应的主要因素是脱水剂的选择,本实验采用EDC·HCl为脱水剂,收率为85%.而影响该工艺路线的关键步骤为化合物IV的脱Boc反应和化合物V的硫代反应.
2.1 脱Boc试剂的选择
脱Boc反应一般使用TFA(三氟乙酸)作为脱Boc试剂[15],同时作为反应溶剂,但是该方法也存在许多问题,如使用大量的TFA(8当量),后处理麻烦,需要加入大量的碱中和,分离过程复杂,产率低,副产物量多,导致产品收率低.本实验中对脱Boc反应中的脱Boc试剂进行优化实验,主要考察了TFA和HCl在不同的反应条件下对反应结果的影响,结果如表1所示.
表1 不同脱Boc试剂对反应结果的影响
Table 1 The effects of Boc-deprotection reagent on the reaction
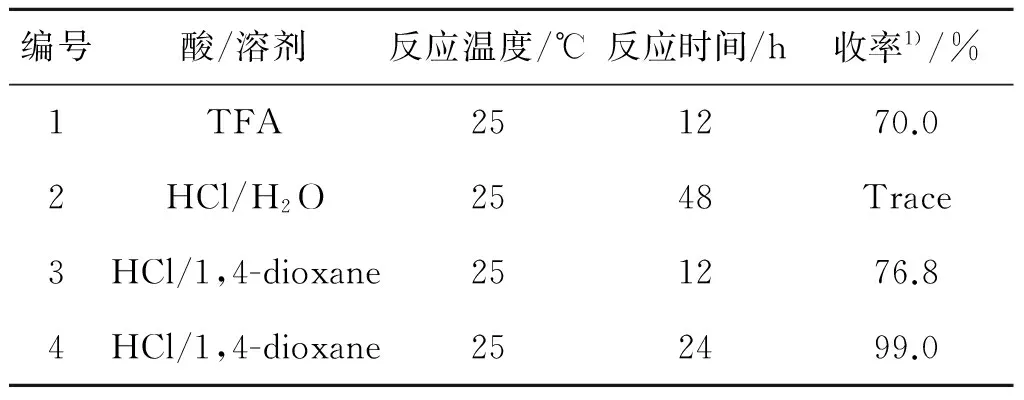
编号酸/溶剂反应温度/℃反应时间/h收率1)/%1TFA251270.02HCl/H2O2548Trace3HCl/1,4-dioxane251276.84HCl/1,4-dioxane252499.0
注:1) 收率均为萃取分离后收率.
实验结果发现,TFA虽然反应速度快,但其产率低,重现性差,伴随生成三氟甲酰胺产物的副反应且该副反应难以避免.采用盐酸水溶液进行脱Boc反应,反应时间长,进行缓慢,持续反应48 h后仍然没有检测到脱Boc产物的生成.而使用1 M的HCl/二氧六环溶液反应12 h即可得到76.8%的收率,延长反应至24 h可得到99%以上的收率,反应效果明显优于TFA,且无副反应发生.而且,该后处理简单,只需进行简单的pH调节和萃取即可得到高收率、高纯度的脱Boc产物.
2.2 溶剂对环化反应的影响
实验中发现,同一反应使用不同的溶剂反应效果相差甚大,溶剂对化合物IV的环化反应具有一定的影响.因此,在反应中如何有效的使用溶剂,己成为一个影响反应结果的问题.在本实验中主要考察了不同溶剂对环化反应结果的影响(表2).

表2 不同溶剂对反应结果的影响
注:1) 收率均为柱层析分离后收率.
L-N-甲基苯丙氨酰胺与丙酮的反应,在甲醇溶剂中反应过夜仅仅获得67%的产率,即使延长反应时间也无明显变化.而采用丙酮为反应溶剂时,反应效果要比使用甲醇做溶剂效果好,即丙酮既作为反应物,又作为反应溶剂,在相同反应时间内,以丙酮为溶剂时反应的产率大于以甲醇为溶剂时的反应收率,柱层析分离后产率达80%,而且可以有效的缩短反应时间到8 h.
2.3 硫代试剂选择
硫代反应是本合成工艺路线的重要步骤之一,常用硫代试剂有硫单质,P2S5,P2S5/Al2O3,P4S10/HMDO,Lawesson试剂等.(S)-5-苄基-2,2-二甲基-3-(咪唑丙基)-4-氧咪唑烷酮V的硫代反应并未见文献报道,本实验就几种简单易得的硫代试剂对化合物V进行了硫代反应的尝试(表3).

表3 不同硫代试剂对反应结果的影响
注:1) 收率均为柱层析分离后收率.
与本课题组之前合成的手性小分子咪唑啉酮相比,本实验合成的(S)-5-苄基-2,2-二甲基-3-(咪唑丙基)-4-氧咪唑烷酮对硫代反应不敏感,P2S5和P2S5/Al2O3能进行常规的咪唑啉酮催化剂的硫代,但无法进行此类取代的咪唑啉酮催化剂的硫代反应,而采用高效的硫代试剂(Lawesson试剂)也仅得到少量产物,反应效果不理想.
根据实验跟踪检测,该实验转化率较高,大部分原料参与了反应,但产物、原料及副产物极性十分相近,在经3~4次柱层析分离后得到部分纯净产物与副产物,其中,化合物VI经3次柱层析分离后的收率仅为22%.经结构鉴定,我们确定该分离得到的副产物结构为产物未脱去Lawesson试剂的中间化合物.根据上述实验结果,我们推测其原因为咪唑啉酮催化剂在侧链上引入一个长链且极性较大的支链后会对咪唑啉酮羰基的反应活性有影响,造成Lawesson试剂无法很好的与羰基结合以及硫代反应后Lawesson试剂的脱除困难,从而阻碍了硫代反应的进行.
3 结 论
本实验以L-苯丙氨酸为手性原料,依次经过Boc保护、酰胺化、脱Boc、环化、硫代、离子化6步反应,制得终产物离子液体负载的手性硫代咪唑啉酮,总收率为12%,ee值为99%.其中,通过对脱Boc反应条件进行优化,采用氯化氢的二氧六环溶液代替三氟乙酸作为脱Boc试剂,获得了较佳的结果.首次开发了取代咪唑啉酮的硫代反应,在此基础上,开发了碘化离子液体负载的手性硫代咪唑啉酮,该离子液体可以在丙酮溶剂中与KBF4或者KPF6进行等当量阴离子交换,从而可制备不同阴离子型离子液体负载的手性硫代咪唑啉酮催化剂.另外,本研究可为新型手性催化剂的开发及应用提供一条可借鉴的研究思路.
[1] 林国强,李月明,陈耀全,等. 手性合成-不对称反应及其应用[M]. 2版. 北京:科学出版社,2005.
[2] SHIBASAKI M, YOSHIKAWA N. Lanthanide complexes in multifunctional asymmetric catalysis[J]. Chemical reviews, 2002, 102(6): 2187-2209.
[3] AHRENDT K A, BORTHS C J,MACMILLIAN D W C.New strategies for organic catalysis: the first highly enantioselective organocatalytic Diels-Alder reaction[J].Journal of the american chemical society,2000,122(17):4243-4244.
[4] WILSON R M,JEN W S,MACMILLIAN D W C.Enantioselective organocatalytic intramolecular Diels-Alder reactions: the asymmetric synthesis of aolanapyrone D[J].Journal of the american chemical society,2005,127(33):11616-11617.
[5] HORNING B D,MACMILLIAN D W C.Nine-step enantioselective total synthesis of (-)-vincorine[J].Journal of the american chemical society,2013,135(17):6442-6445.
[6] JEN W S,WIENER J J M,MACMILLIAN D W C.New strategies for organic catalysis: the first enantioselective organocatalytic 1,3-dipolar cycloaddition[J].Journal of the american chemical society,2000,122(40): 9874-9875.
[7] PARAS N A,MACMILLIAN D W C.New strategies in organic catalysis:the first enantioselective organocatalytic Friedel-Crafts alkylation[J].Journal of the american chemical society,2001,123(18):4370-4371.
[8] AUSTIN J F,MACMILLIAN D W C.Enantioselective organocatalytic indole alkylations: design of a new and highly effective chiral amine for iminium catalysis[J].Journal of the american chemical society,2002,124(7):1172-1173.
[9] ALLEN A,MACMILLIAN D W C.Enantioselective α-arylation of aldehydes via the productive merger of iodonium salts and organocatalysis[J].Journal of the american chemical society,2011,133(12):4260-4263.
[10] GUALANDI A,EMER E,CAPDEVILA M G.Highly enantioselective an alkylation of aldehydes with 1,3-benzodithiolylium tetrafluoroborate: a formal organocatalytic a alkylation of
aldehydes by the carbenium Ion[J].Angewandte chemie international edition,2011,50(34):7842-7846.
[11] CHIARUCCI M,LILLO M D,BANDINI M..Gold meets enamine catalysis in the enantioselective α-allylic alkylation of aldehydes with alcohols[J].Chemical science,2012,3(9):2859-2863.
[12] COMITO R J,FENILLI F G,MACMILLIAN D W C.Enantioselective intramolecular aldehyde α-alkylation with simple olefins: direct access to Homo-ene products[J].Journal of the american chemical society,2013,135(25):9358-9361.
[13] LIANG X R,FAN J Y,SHI F,et al.Imidazolethiones: novel and efficient organocatalysts for asymmetric Friedel-Crafts alkylation[J].Tetrahedron letters,2010,51(18):2505-2507.
[14] LIANG X R,LI S M,SU W K.Highly stereoselective imidazolethiones mediated Friedel-Crafts alkylation of indole derivatives[J].Tetrahedron letters.2012,53(3):289-291.
[15] LIANG X R,LI N,CHEN X L,et a1.Asymmetric α-oxyamination of aldehydes by synergistic catalysis of imidazolethiones and metal salts[J].RSC advances,2014,4(83):44039-44042.
[16] LI N,LIANG X R,SU W K.New insights into the asymmetric Diels-Alder reaction: the endo-and S-selective retro-Diels-Alder reaction[J].RSC advances,2015,5(128):106234-106238.
[17] SHEN Z L,CHEONG H L,LAI Y C,LOH T P.Application of recyclable ionic liquid-supported imidazolidinone catalyst in enantioselective Diels-Alder reactions[J].Green chemistry,2012,14(9):2626-2630.
[18] 毛信表,任淑鹰,张寅旭,等.CO2在[bmim]BF4/[bmim]PF6中的电化学还原研究[J].浙江工业大学学报,2014,42(6):636-639.
[19] 欧阳启,黄金,王普,等.离子液体对热带假丝酵母细胞生物相容性的研究[J].浙江工业大学学报,2013,41(3):254-259.
[20] 王普,任峰,黄金,等.含离子液体介质中生物拆分制备(S)-(+)-2,2-二甲基环丙烷甲酸[J].浙江工业大学学报,2013,41(4):409-414.
[21] SHEN Z L,KAU K K G.,LOH T P.Synthesis and application of a recyclable ionic liquid-supported imidazolidinone catalyst in enantiselective 1,3-dipolar cycloaddition[J].Chemical communications,2012,48(47):5856-5858.
(责任编辑:刘 岩)
A novel synthesis method of (S)-type ionic liquid immobilized imidazolethinones
LIANG Xianrui1,2, CHEN Xinlei1,2
(1.Collaborative Innovation Center of Yangtze River Delta Region Green Pharmaceuticals, Zhejiang University of Technology,Hangzhou 310014, China;2.College of Pharmaceutical Sciences, Zhejiang University of Technology, Hangzhou 310014, China)
A novel synthesis method of (S)-type ionic liquid immobilized imidazolethinones was reported in this paper.(S)-3-(3-(1H-Imidazol-1-yl)propyl)-5-benzyl-2,2-dimethylimidazolidin-4-one was synthesized from L-phenylalanine via Boc-protection, amidation, Boc-deprotection, cyclization. Then it was followed the sulfurated reaction with using Lawesson reagent as sulfuration agent in toluene at 80 ℃ for 8 hrs to give the chiral imidazolethinone in high ee value. The reaction of the obtained imidazolethione with n-iodobutane produced the (S)-type ionic liquid immobilized imidazolethinone. The Boc-deprotection product was obtained in quantitative yield by using the HCl/1,4-dioxane as reaction reagents.
imidazolethione; N-Boc deprotection; ionic liquid
2016-03-15
国家自然科学基金资助项目(21206148)
梁现蕊(1975—),女,山东临沂人,副教授,博士,研究方向为手性合成及药物分析,E-mail:liangxrvicky@zjut.edu.cn.
O622
A
1006-4303(2016)05-0510-05
猜你喜欢
分子催化(2022年1期)2022-11-02
农业工程学报(2022年13期)2022-10-09
科学导报(2022年41期)2022-07-13
功能材料(2022年5期)2022-06-02
中华养生保健(2020年3期)2020-11-16
地球科学与环境学报(2020年2期)2020-03-26
农药科学与管理(2019年9期)2019-11-23
浙江化工(2019年3期)2019-04-09
分析化学(2016年11期)2017-04-25
肿瘤影像学(2015年3期)2015-12-09
