
基于等离子体电解氧化的铝铜合金表面陶瓷化处理
2016-11-11 03:46王达成江开勇
海峡科学 2016年7期
王达成 江开勇 赵 迪
基于等离子体电解氧化的铝铜合金表面陶瓷化处理
王达成 江开勇 赵 迪
华侨大学
该文利用等离子体电解氧化技术(PEO)对铝铜合金表面进行高效陶瓷化处理,利用自制的等离子体电解氧化装置对铝铜合金材料的等离子体电解氧化行为及生成的陶瓷层特性进行研究,利用扫描电镜分析了陶瓷层的形貌和组织结构。
等离子体电解氧化 铝铜合金 电源参数 致密性
等离子体电解氧化(Plasma Electrolytic Oxidation)又称微弧氧化(MAO)、微弧放电氧化(MDO)、火花放电阳极氧化(ANOF)或阳极火花沉积(ASD)[1-5],可在轻金属(Al、Mg、Ti等)材料表面原位生长出耐蚀、耐磨的氧化陶瓷膜。该技术处理所得氧化膜与金属基体间的结合力强,膜厚范围较宽且可控,形成的复合材料具有高硬度、耐腐蚀、耐磨损、抗热震等优异性能。目前,美、俄、日、德等国已将PEO应用于尖端武器装备的制造中,解决了许多其他方法无法解决的关键技术问题。
铝铜合金是指以铜为主要合金元素的铝合金,它包括Al-Cu-Mg合金、Al-Cu-Mg-Fe-Ni合金和Al-Cu-Mn合金等,属热处理可强化合金。其特点是强度高,通常称为硬铝合金,耐热性能和加工性能良好,但耐蚀性不如大多数其他铝合金好,在一定条件下会产生晶间腐蚀,因此板材往往需要包覆一层纯铝或6系列铝合金,提高抗腐蚀性。陶瓷材料以其特有的耐磨、耐腐蚀、耐高温等性能,以及丰富的资源优势,被称为继钢铁、铝材后的第三代工程材料。由于陶瓷材料脆性大,可加工性差,未能得到广泛使用。合金表面陶瓷化的研究目的在于,在保证使用性能的前提下,用廉价的金属代替贵重的技术材料,并克服陶瓷材料难以加工的问题,从而提高陶瓷材料的可成型性和可加工性,同时赋予材料一些其它表面强化技术无法得到的特殊性能,拓展陶瓷材料的使用范围和应用领域。铝合金表面陶瓷化技术可应用于铝合金建筑材料、工业型材、建筑板材、活塞、电子工业用特种零部件的处理,其市场前景极为广阔。
1 等离子体电解氧化电源
本文使用的等离子体电解氧化陶瓷层的制备系统是自制的100W等离子体电解氧化装置。装置的电源输出频率可调,额定输出电压为3kV,电流为30mA。装置电源的输出波形如图1所示。
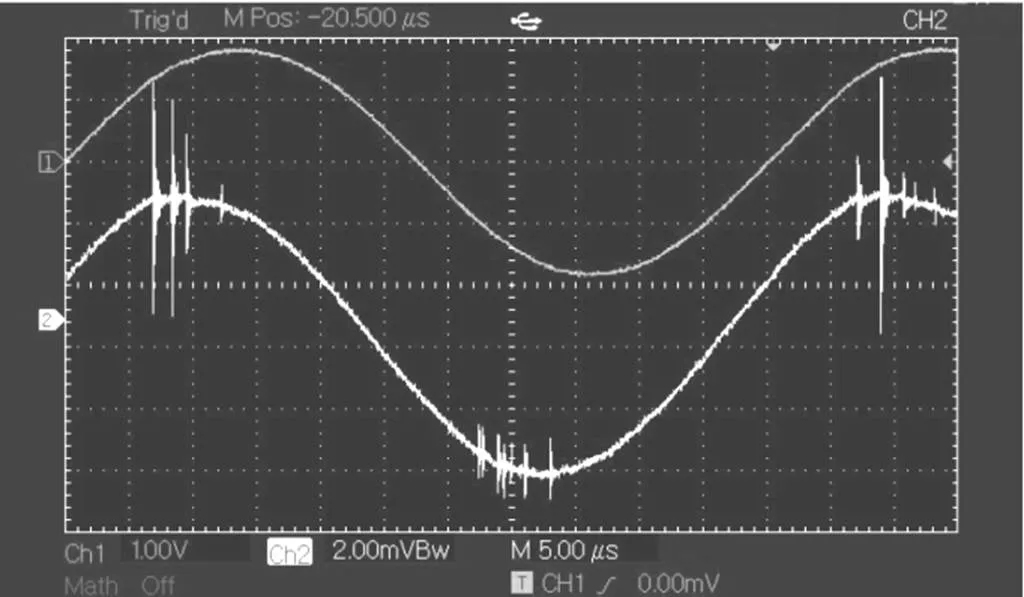
图1 等离子体电源输出波形
2 等离子体电解氧化实验装置
本文采用的等离子体电解氧化装置如图2所示。它包括等离子体电解氧化电源、电解槽、恒温磁力搅拌器、循环冷却系统和测温系统。电解槽为玻璃容器,处理式样作为阳极。整个等离子体电解氧化过程中,为保持处理液的浓度和温度各处均匀,使用恒温磁力搅拌器来进行加速传质。处理液的温度在实验过程中保持在35oC以下,以避免因溶液蒸发而改变处理液的浓度。
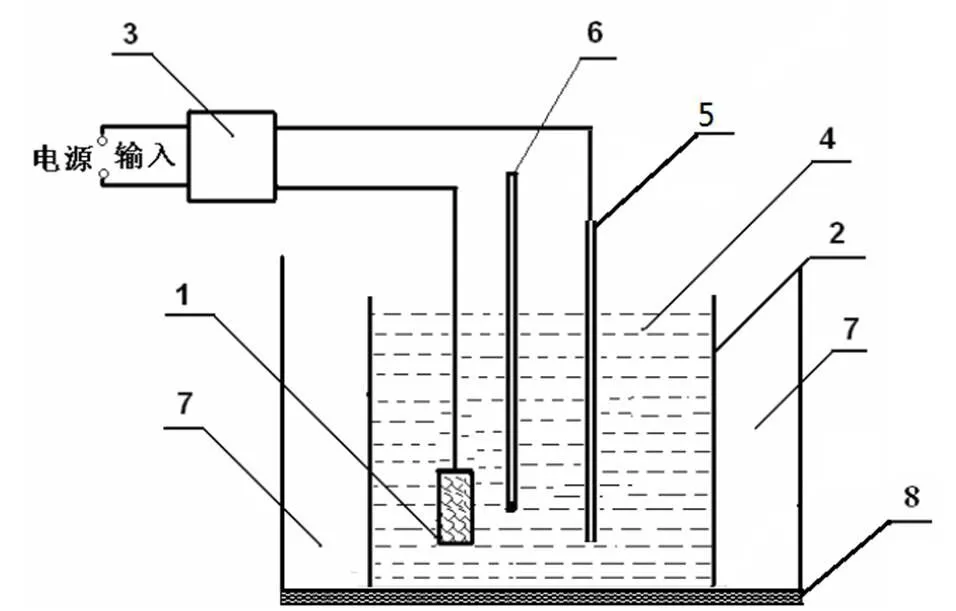
1——试样;2——电解槽;3——电源;4——处理液;5——阴极;6——温度计;7——冷却水循环系统;8——恒温磁力搅拌器
3 实验材料和处理工艺
本实验用到的铝质材料为LY12硬铝合金材料,强度高,有一定的耐热性,可用作150°C以下的工作零件。温度高于125°C,2024合金的强度比7075铝合金还高。热状态、退火和新淬火状态下成形性能都比较好,热处理强化效果显著,但热处理工艺要求严格。抗蚀性较差,但用纯铝包覆可以得到有效保护;焊接时易产生裂纹,但采用特殊工艺可以焊接,也可以铆接。广泛用于飞机结构、铆钉、卡车轮毂、螺旋桨元件及其他结构件。
等离子体电解氧化工艺简单,不需酸洗、活化等复杂的前处理过程。实验中所用到的高纯铝试样,表面已经抛光好,不需要打磨,铝铜合金处理前,需用SiC砂纸对试样进行打磨,最后一道砂纸为800#。打磨好的合金和高纯铝试样依次用丙酮、酒精和去离子水清洗,然后吹干备用。处理完的试样只需用自来水冲去表面残留的处理液,然后用热风机吹干、编号,以备检测之用。
4 研究方法
4.1 膜层结构和成分分析
使用HITACH S-4200型扫描电子显微镜观察陶瓷层的表面及断面形貌。观察断面形貌时,由于陶瓷层较脆,砂纸打磨时易崩落,而且铝层较软,容易堆边出现倒角,因此预先用配置好的环氧树脂 (100 g环氧树脂+5~8 mL乙二胺) 对试样镶嵌。试样镶嵌完毕并固化之后,用砂纸逐级打磨,最后一道砂纸为1200#,然后用Cr2O3抛光液抛光至观察表面无划痕,清洗干净后,用15%的NaOH溶液进行腐蚀,用酒精擦干,待观察。由于所制得的陶瓷层不导电,为防止观察时的表面放电现象,在进行表面及截面形貌观察前,需要对试样表面进行喷金处理,并使用扫描电子显微镜配置的能谱仪进行成分的点、线和面分析,研究膜层内部各种元素的分布特点。
4.2 陶瓷层的表面和断面微观结构
图3为处理过程1#、2#、4#和5#中陶瓷层的表面形貌。从中清晰看到由于击穿和重新凝固而形成的残留放电通道和盘状物,而且可以明确了解不同生长条件下陶瓷层表面形貌的差异。
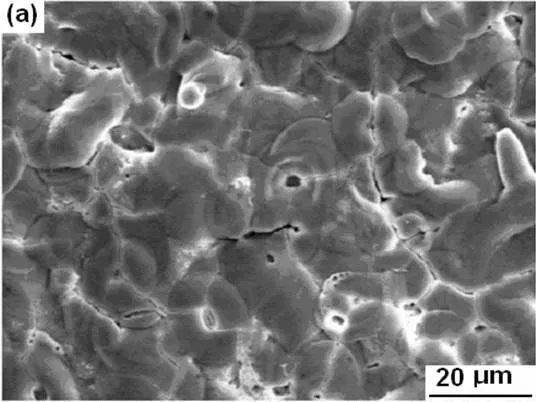
(a)SI体系30 min (b)P体系30 min (c)P体系30 min,然后SI 体系30 min (d)SI体系30 min,然后P体系30 min
图3(a)显示,在SI体系中制得的陶瓷层具有较为均匀的表面形貌,可以发现许多尺寸为1~3 μm不等的微孔,然而在P体系获得的陶瓷层表面的孔洞平均尺寸则大于8 μm,有些部位甚至有几个孔洞贯通连接在一起的形貌出现,而且陶瓷层表面显得略为粗糙,有一些微小的裂纹存在,如图3(b)所示。在图3(b)中,还可以看到较为明显的盘状物,这些盘状物的尺寸要大于图3(a)中盘状物。比较有意思的是,P体系中获得的陶瓷层在SI体系中继续处理以后,图3(b)中显示的较大尺寸的孔洞有所减小,变为直径5 μm左右,微裂纹的数量也有所减少,具体微观形貌如图3(c)所示。同样,图3(d)显示,SI体系中获得的陶瓷层在P体系中继续处理以后,陶瓷层表面的孔洞数量和尺寸明显变大,很多贯通的孔洞开始出现,而且陶瓷层表面的粗糙度相对于图3(a) 和 (c)明显变大。
图3所示的陶瓷层不同形貌与不同的等离子体电解氧化过程有很大关系。对于P体系处理液而言,由于(NaPO3)6是一种环状聚合物,因此在反应中需要更大的能量去打开分子之间的结合[6],P体系中的电压要稍微高一些。此外,相同浓度条件下,P体系处理液的电导率要低于SI体系处理液,这也是导致P体系中的电压稍微偏高的原因[7]。高的输入能量势必造成试样表面相对更为强烈的放电现象,放电通道内形成的大量熔融物质在电场力的作用下从放电通道喷出,因此P体系中形成的陶瓷层具有大尺寸的放电孔洞和盘状结构。
对应上述不同实验过程所获得的陶瓷层表面形貌,图4给出了不同处理液体系中制得陶瓷层的断面形貌。
如图4所示,P体系中所获得的陶瓷层断面结构最为疏松,可以看到许多较大的裂纹和孔洞,这与图3.2(b)中所显示出来的较为粗糙的表面结构是联系在一起的。此外还可以看出,再次经过SI体系进行处理,虽然陶瓷层略变致密,但是在陶瓷层和基体的界面附近仍可发现较为明显的组织缺陷。相对于图4(b)和图4(c),图4(a)和图4(d)的断面结构显得致密的多。这种断面结构同样与P体系放电电压高、放电剧烈有密切关系。

(a)SI体系30 min (b)P体系30 min (c)P体系30 min,然后SI 体系30 min (d)SI体系30 min,然后P体系30 min
4.3 关于离子跟踪实验的讨论
众所周知,等离子体电解氧化反应是一个复杂的物理化学反应过程,在高电压和电流密度条件下,放电通道内部可能发生化学氧化、电化学氧化以及等离子体化学氧化等诸多反应,处理液除了作为反应环境以外,还直接参与上述反应。先前的许多关于传统阳极氧化的研究也都涉及到了处理液组分参与氧化反应的过程[8, 9]。研究表明,在传统的阳极氧化反应中,能够进入陶瓷层的处理液的组分可能带正电荷或者负电荷,也有可能呈电中性,这些组分的运动方向也是多种多样。有的在电场的作用下向陶瓷层内部或者外侧运动,而有的可能静止不动。那些向陶瓷层内部运动的离子或者离子团被包裹在整个通道的各处,而向外侧运动的组分则主要分布在陶瓷层和处理液的界面。我们认为,这种分布规律同样适用于等离子体电解氧化过程。
等离子体电解氧化过程中,电解液中的离子在电泳作用下到达膜层与电解液表面。电击穿在已经生成的膜层中产生低电阻通道,电解液进入到通道内部,在高温下与熔化了的基底以及原有的膜层发生反应,生成新的膜层。这期间,电解液中的阳离子由于其带正电,大部分会通过膜层孔洞和裂纹排出,所以放电通道内主要是阴离子与熔融的基底相互作用,在高温下生成晶态化合物。当前实验过程中主要有如下反应:

(2)
(3)
如式(2)所示,由于放电通道边缘熔融氧化铝的存在,说明其周围温度必然高于2045℃,而且单个通道的火花电流密度高达2.1×104A·cm-2[10],这样高的能量条件足以引起硅酸盐电解形成SiO2。所以SiO32-离子(当然电解液中也会含有HSiO3-,但HSiO3-不稳定,在高电场下很容易分解)在放电通道内反应生成了具有高结合能的SiO2[8,11]。而处理液和陶瓷层界面的溶胶层会阻止SiO2进入放电通道内部[12],所以大部分SiO2在膜层外部沉积下来,另外,SiO2与熔融的Al2O3进而形成Al-Si-O相,这可以在图4中得到验证,Al-Si-O相同样也会沉积在膜层外部,而Si元素主要分布在陶瓷层外侧。
对于P体系处理液,由于水解反应产生的各种磷酸根(PO43-, HPO42-等)均携带负电荷,在高压电场的驱动下,它们很容易进入放电通道的内部,放电熄灭后,在熔质重新凝固的过程中被包裹起来,成为陶瓷层的一部分,因此可以在陶瓷层的内部发现Al-P-O的存在。
5 结论
我们选取硅酸盐、磷酸盐以及硅酸盐和磷酸盐的混合电解液体系,研究了溶液离子在等离子电解氧化陶瓷层中的分布规律,建立了等离子体电解氧化过程中的离子输运模型。我们还测得了浸泡不同时间的等离子体电解氧化陶瓷膜的电化学阻抗谱,分析了膜层的结构信息及保护失效过程,具体结论如下:
(1)等离子体电解氧化过程中处理液的组分也参与到反应当中,并可成为陶瓷层的一部分。
(2)在硅酸盐、磷酸盐以及混合体系中进行等离子体电解氧化,发现在磷酸盐中,氧化过程的放电电压高,从而形成的高温稳定相α-2O3的含量也高。
(3)不同的处理液组分由于其不同的运动规律,在陶瓷层中的分布具有不同的特点。以硅酸盐和磷酸盐体系中所制得的陶瓷层为例,Si元素的分布大部分集中在陶瓷层外部,而P元素在整个陶瓷层的断面结构中均有分布。
(4)等离子体电解氧化陶瓷层由外部疏松层和内部致密层组成。腐蚀开始时,陶瓷层的阻值很大,对基底起到保护作用。随着腐蚀过程的进行,电解液将先渗透到陶瓷层内部致密层,在这过程中,腐蚀产物会堵塞电解液渗入的通道,延缓电解液渗透,从而产生扩散现象。最后,陶瓷层内部致密层因腐蚀被破坏,电解液到达膜层与基底界面,陶瓷层保护失效,金属基底开始发生腐蚀。
参考文献:
[1] Long B.H., Wu H.H., Long B.Y., et al. Characteristics of electric parameters in aluminium alloy MAO coating process[J]. J.Phys.D: Appl. Phys, 2005, 38(18): 3491-3496.
[2] Voevodin A. A., Yerokhin A. L., Lyubimov V. V., et al. Charac terization of wear protective Al---Si---O coatings formed on Al-based alloys by micro-arc discharge treatment[J]. Surf. Coat. Technol, 1996, 86(1): 516-521.
[3] Xin S.G., Song L.X., Zhao R.G., et al. Properties of aluminium oxide coating on aluminium alloy produced by micro-arc oxidation[J]. Surf. Coat. Technol, 2005, 199(2): 184-188.
[4] Liu F., Wang F.P., Shimizu T., et al. Formation of hydroxyapatite on Ti-6Al-4V alloy by microarc oxidation and hydrothermal treatment[J]. Surf. Coat. Technol, 2005, 199(S2/3): 220-224.
[5] Guo H.F., An M.Z., Xu S., et al. Formation of oxygen bubbles and its influence on current efficiency in micro-arc oxidation process of AZ91D magnesium alloy[J]. Thin Solid Films, 2005, 485(112): 53-58.
[6] 李建中,邵忠财,田彦文,等.不同含磷处理液在等离子体电解氧化过程中的作用[J].中国腐蚀与防护学报, 2004, 24(4):222-225.
[7] Liang J., Hu L.T., Hao J.C.. Characterization of microarc oxidation coatings formed on AM60B magnesium alloy in silicate and phosphate electrolytes[J]. Appl. Surf. Sci., 2007, 253(10): 4490-4496.
[8] Wood G. C., Skeldon P., Thompson G. E., et al. A model for the incorporation of electrolyte species anodic alumina[J]. J. Electrochem. Soc., 1996, 143(1):74-83.
[9] Skeldon P., Shimizu K., Thompson G.E., et al. Selective interfacial processes and the incorporation of electrolyte species into anodic film on aluminum[J]. Philosophical Magazine B, 1995, 72(4): 391-400.
[10] Van T.B., Brown S.D., Wirtz G.P.. Mechanism of anodic spark deposition[J]. Am. Ceram. Soc. Bull, 1977, 56(6): 563-566.
[11] Monfort F., Berkani A., Matykina E., et al. A tracer study of oxide growth during spark anodizing of aluminum[J]. J. Electrochem. Soc. 2005, 152(6): C382-C387.
[12] Lv G.H., Gu W.C., Chen H., et al. Characteristics of oxide coatings on aluminum by plasma electrolytic oxidation in silicate and phosphate electrolyte[J]. Appl. Surf. Sci, 2006, 253(5): 2947-2952.
猜你喜欢
电子技术与软件工程(2021年7期)2021-06-16
中学生数理化(高中版.高考理化)(2020年3期)2020-05-30
山东冶金(2019年5期)2019-11-16
收藏界(2019年3期)2019-10-10
制造技术与机床(2017年12期)2017-02-02
光学精密工程(2016年4期)2016-11-07
现代工业经济和信息化(2016年5期)2016-05-17
电源技术(2016年9期)2016-02-27
中国资源综合利用(2016年7期)2016-02-03
应用化工(2014年1期)2014-08-16
