
锰(Ⅱ)-纳米银体系共振散射光谱法检测痕量过氧化氢
2016-11-08 11:12唐宁莉陈永宁张容珲覃温露
分析测试学报 2016年9期
唐宁莉,陈永宁,张容珲,覃温露
(桂林理工大学 化学与生物工程学院,广西 桂林 541004)
锰(Ⅱ)-纳米银体系共振散射光谱法检测痕量过氧化氢
唐宁莉*,陈永宁,张容珲,覃温露
(桂林理工大学化学与生物工程学院,广西桂林541004)
利用纳米银(AgNPs)作为共振散射探针,根据反应前后纳米银聚集状态改变所引起的体系的共振散射信号的变化,对H2O2进行定量检测。采用硼氢化钠还原法,在柠檬酸三钠保护下将银从硝酸银中还原,合成了平均粒径约为4 nm的银纳米微粒。Mn2+和覆盖在纳米银表面的柠檬酸根负离子络合诱导纳米银聚集,导致体系的共振散射强度增强;当体系中存在H2O2时,H2O2和Mn2+发生类Fenton反应生成Mn3+和羟基自由基(·OH)。Mn3+和纳米银不能形成稳定的络合物,·OH氧化蚀刻纳米银,两种反应共同导致纳米银的聚集程度减弱,体系的共振散射信号强度相应减弱,据此建立了一种检测痕量H2O2的共振散射光谱新方法。在最佳条件下,波长411 nm处,体系的共振散射强度变化值(ΔI)与H2O2浓度在7.88×10-8~1.418×10-5mol/L范围内呈线性关系,相关系数为0.996 7,检出限为4.04×10-8mol/L。该方法应用于湖水和雨水中H2O2的检测,结果满意。
过氧化氢;纳米银;共振散射;类Fenton反应
过氧化氢(H2O2)是一种常见且常用的消毒剂和漂白剂,被广泛应用于食品、制药、临床医学、有机合成、环境分析等行业。另一方面,H2O2参与人体细胞免疫和细胞信号的转换过程,是人体细胞内葡萄糖的氧化代谢产物[1]。然而,空气中存在的H2O2与酸雨的形成有着密切关系,对环境破坏起到一定的推动作用[2];H2O2也是一种强氧化剂,在生物体内若不能及时分解,则会瓦解细胞膜,使机体部分功能丧失。人体内如果存在过多的H2O2,H2O2将会转变成毒性的·OH从而加速人体老化,降低生育率,并对人体造成严重伤害[3]。因此,H2O2含量的测定显得尤为重要。
目前测定H2O2的方法有分光光度法[4-6]、荧光光度法(FS)[7-9]、高效液相色谱法(HPLC)[10-11]、电化学分析法(EC)[12-15]、化学发光法(CL)[16]、共振散射光谱法(RLS)[17-19]和比色法[20-21]等。共振散射光谱法因具有简便快速、灵敏度高、检出限低等特点而引起了广大分析工作者的关注。纳米技术是一项新兴的科学技术,基于金属纳米粒子特殊的表面性质,纳米金属已被应用于各个领域[22-23]。纳米银因具有独特的局部表面等离子共振(LSPR)特性而被应用于一些物质的共振散射光谱分析[24]。本文研究了纳米银与Mn2+的络合反应以及Mn2+和H2O2的类Fenton反应,并建立了一种简单快速、高灵敏测定痕量H2O2的共振散射光谱新方法。
1 实验部分
1.1 仪器与试剂
Cary Eclipse荧光分光光度计、Cary 50紫外-可见分光光度计(美国Varian公司);HH-42快速恒温数显水箱(常州国华电器有限公司);PHS-3C 精密pH计(上海雷磁仪器厂)。
H2O2储备溶液:取2 mL 30%(体积分数) 的H2O2于500 mL容量瓶中,用超纯水定容至刻度,用高锰酸钾标定法测得H2O2储备溶液浓度为3.94×10-2mol/L,H2O2工作溶液(7.88×10-5mol/L)临用前由储备液用超纯水稀释得到;氯化锰(MnCl2):1.0×10-3mol/L;硝酸银:1.0×10-3mol/L;硼氢化钠:2.0×10-3mol/L;柠檬酸三钠:10 g/L;醋酸-醋酸钠(HAc-NaAc)缓冲液:将0.1 mol/L醋酸溶液与0.1 mol/L醋酸钠溶液混合,调至pH 4.0~5.5;Clark-Lubs(C-L)缓冲液:将0.1 mol/L邻苯二甲酸氢钾溶液和0.1 mol/L氢氧化钠溶液混合,调至pH 4.0~5.5;Britton-Robinson( B-R)缓冲液:由浓度均为0.04 mol/L的醋酸、硼酸、磷酸溶液等体积混合,用0.2 mol/L氢氧化钠溶液调至pH 4.0~5.5。所用试剂均为分析纯,实验用水为超纯水。
1.2 实验方法
1.2.1纳米银的合成 纳米银参考文献[25]稍作修改合成:室温下,在剧烈搅拌(750 r/min)的75 mL 2.0×10-3mol/L硼氢化钠溶液中逐滴匀速地加入25 mL 1.0×10-3mol/L的硝酸银溶液,持续搅拌10 min后,快速加入5 mL 10 g/L的柠檬酸钠溶液,以稳定胶体。胶体继续搅拌20 min后装入棕色容量瓶,放入冰箱(4 ℃)保存孵化2 d后待用,胶体中银纳米微粒的浓度为2.5×10-4mol/L。
1.2.2过氧化氢的测定 在10 mL比色管中,依次加入1.0 mL HAc-NaAc缓冲溶液(pH 5.2)、150 μL 1.0×10-3mol/L的MnCl2溶液与一定量的H2O2工作液,室温下放置20 min后,加入1.0 mL 2.5×10-4mol/L的纳米银溶液,用水定容至10.0 mL,反应5 min后,在荧光光度计上以λem=λex(Δλ=0 nm)进行同步扫描(狭缝2.5 nm,电压650 V),测量波长411 nm处反应体系的共振散射强度I,同时以不加H2O2体系的共振散射强度作为空白I0,计算ΔI=I0-I。
2 结果与讨论
2.1体系的紫外-可见吸收光谱

图1 体系的紫外-可见吸收光谱Fig.1 UV-Vis absorption spectra of different systemsa.MnCl2;b-g.MnCl2+AgNPs+(0,0.245, 1.636,3.068,4.908,6.953) μmol/L H2O2;h.AgNPs;[MnCl2]:1.5×10-5 mol/L,[AgNPs]:2.5×10-5 mol/L;insert:relation of absorbance changed value ΔA and concentration of H2O2
体系的紫外-可见吸收光谱见图1(插图为吸光度差值ΔA与H2O2浓度的线性关系)。由图1可知,纳米银在398 nm附近有明显的吸收(图1曲线h),而MnCl2几乎无吸收(图1曲线a)。当向纳米银溶液中加入MnCl2溶液后,纳米银的吸收峰急剧下降(图1曲线b),说明此时纳米银和MnCl2发生了反应。当MnCl2和H2O2同时存在时,纳米银的吸收峰下降程度减弱,且随着H2O2浓度的增大,纳米银吸收峰的降低越不明显(图1曲线c~g),说明H2O2的存在阻碍了纳米银和MnCl2的反应。

图2 体系的共振散射光谱Fig.2 Resonance light scattering(RLS)spectra of systemsa.MnCl2+ AgNPs;b-i.MnCl2+AgNPs+(0.078 8,1.58,3.94,6.30,7.88,9.46,11.82,14.18) μmol/L H2O2;[MnCl2]:1.5×10-5 mol/L,[AgNPs]:2.5×10-5 mol/L;insert:calibration curve of H2O2
2.2体系的共振散射光谱
体系的共振散射光谱如图2所示。在pH 5.2的HAc-NaAc缓冲溶液中,Mn2+的作用下,纳米银发生聚集,其共振散射信号增大,最强散射峰位于411 nm处(图2曲线a)。当体系中继续加入H2O2时,纳米银的聚集程度相比加入H2O2前有所减弱,产生的共振散射信号随之减弱,且共振散射强度随着H2O2加入量的增加而减小,减弱程度与H2O2加入量呈一定的线性关系(图2曲线b~i)。
2.3反应机理
实验合成的纳米银以小颗粒(约4 nm)状态均匀分布在溶液中(图3A),其表面覆盖着大量的柠檬酸根。具有3d轨道的Mn2+为5配位或6配位,—COOH与金属离子之间具有极强的吸引力[26-27],因此Mn2+不仅可与任意一个纳米银表面带—COOH的柠檬酸根通过金属配位作用络合,也可与邻近纳米银表面上的柠檬酸根络合,从而降低了纳米银表面柠檬酸根的覆盖密度,减少了纳米银表面的负电荷,并导致纳米银大量聚集[28](图3B)。
大多数过渡金属离子与H2O2可发生Fenton或类Fenton反应,反应方程式为:
产物中的金属阳离子以不稳定的氧化态存在[29]。Mn2+与H2O2可发生类Fenton反应,生成不稳定的Mn3+[30-31],反应方程式为:
文献表明,Mn3+对H2O2具有更强烈的催化分解作用[31];Fenton或类Fenton反应产生的·OH可以氧化纳米银[32]。故当纳米银与Mn2+配位络合,同时存在H2O2时,发生类Fenton反应,生成Mn3+,使Mn2+浓度不断降低,而生成的Mn3+与纳米银表面的柠檬酸根不能形成稳定络合物;同时银纳米微粒不断被·OH氧化蚀刻,其浓度降低,这两种反应的结果使得纳米银与Mn2+络合后的聚集程度减弱(图3C),体系的共振散射强度则随H2O2的加入而减弱,所得透射电子显微镜(TEM)结果与共振散射光谱结果相一致。
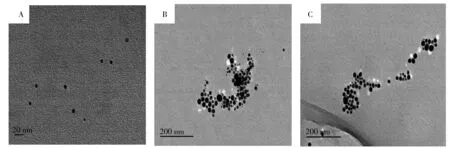
图3 体系的透射电子显微镜图Fig.3 TEM images of systemsA.AgNPs;B.MnCl2+ AgNPs;C.MnCl2+ AgNPs+ H2O2;[MnCl2]:1.5×10-5 mol/L,[AgNPs]:2.5×10-5 mol/L,[H2O2]:4.728 μmol/L
据此本文提出了Mn2+-AgNPs-H2O2体系的反应机理(如图4)。
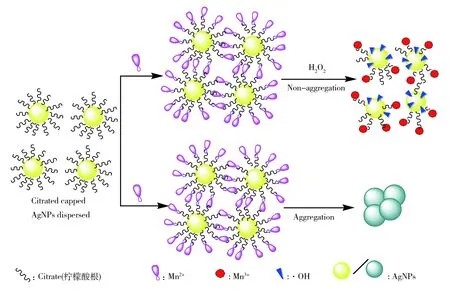
图4 体系的反应机理Fig.4 The reaction mechanism of systems
2.4反应条件的选择
2.4.1缓冲溶液的选择分别考察了B-R、HAc-NaAc和C-L缓冲溶液对体系ΔI值的影响。结果显示,在HAc-NaAc缓冲溶液(pH 5.2)中,体系的ΔI值最大,反应最灵敏,故以HAc-NaAc缓冲溶液(pH 5.2)作为反应的最佳介质。
2.4.2缓冲溶液用量的选择比较了HAc-NaAc缓冲溶液(pH 5.2)不同用量条件下体系ΔI值的大小。结果表明,当缓冲溶液的用量为1.0 mL时,ΔI值最大,故实验选择加入1.0 mL的HAc-NaAc缓冲溶液。
2.4.3MnCl2用量的选择考察了MnCl2用量对体系ΔI值的影响。结果表明,MnCl2用量为150 μL时,体系的ΔI值最大,当MnCl2用量大于150 μL时,ΔI值随用量的增加反而减少,故选择加入MnCl2溶液150 μL。
2.4.4纳米银用量的选择 考察了纳米银用量对体系ΔI值的影响。结果表明,当纳米银用量为1.0 mL时,体系的ΔI值达到最大,随着纳米银用量的继续增大,体系ΔI值减小,因此纳米银的最佳用量为1.0 mL。
2.4.5反应时间的选择考察了反应时间对体系ΔI值的影响。使MnCl2和H2O2反应20 min后再加入AgNPs,体系ΔI值达到最大且最稳定。加入AgNPs后在1~100 min内体系稳定。故实验选择在MnCl2和H2O2反应20 min后加入AgNPs,并于定容5 min后测定。
2.5标准曲线与方法检出限
在优化实验条件下,测定含不同浓度(0.078 8,1.58,3.94,6.30,7.88,9.46,11.82,14.18 μmol/L)H2O2的反应体系的共振散射强度值。在波长411 nm处,H2O2浓度(c,mol/L)在7.88×10-8~1.418×10-5mol/L范围内与体系ΔI值呈良好线性关系,回归方程为ΔI=1.780×107c+57.42,相关系数(r)为0.996 7。按照实验方法平行测定空白体系11次,标准偏差S=0.24,相对标准偏差(RSD)为0.059%,根据DL=3S/K求得方法检出限(DL)为4.04×10-8mol/L。与已报道的方法相比,本方法所用试剂种类少,操作简便,线性范围宽,灵敏度高(见表1)。
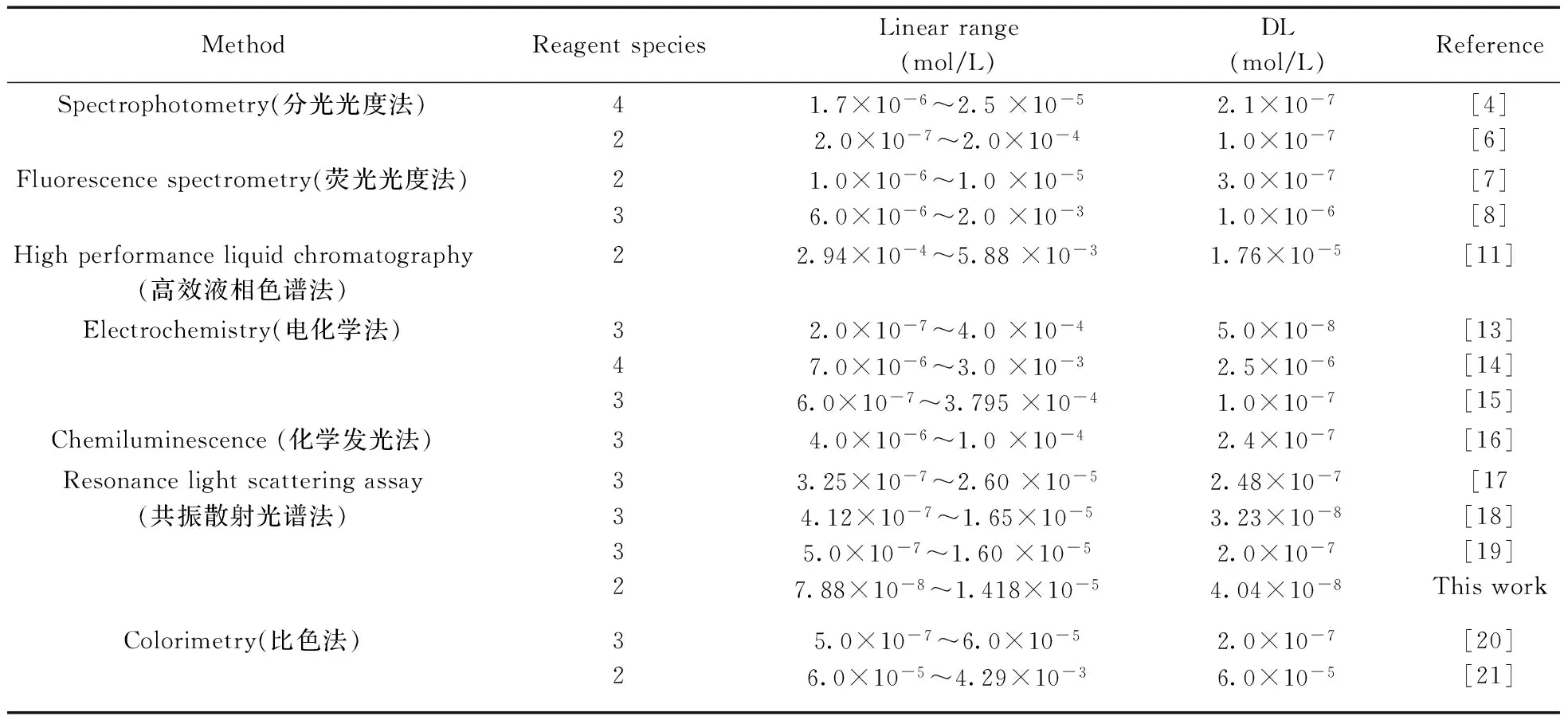
表1 过氧化氢测定方法的比较Table 1 Comparison of some assays for H2O2
2.6干扰物质的影响
按实验方法,当H2O2浓度为3.94×10-6mol/L时,考察了共存物质(CS)的干扰情况,当相对误差的≤± 5%时,共存物质的允许量见表2。由表2可知,除了少量金属离子(Cu2+,Pb2+,Fe3+等)对体系的干扰较大外,其他常见物质的允许浓度较大,对H2O2的测定几乎无影响,方法的选择性较好。
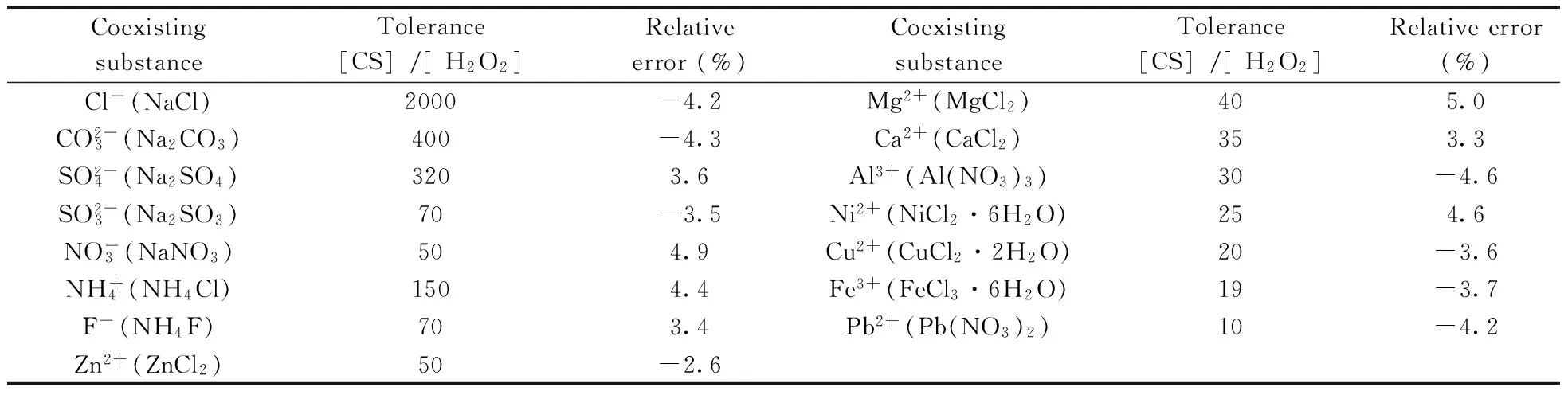
表2 共存物质对体系的影响Table 2 Effect of coexisting substances on system
2.7样品分析与回收率实验
采集学校校园内湖水和新鲜雨水水样各1份,经过滤处理后,静置10 min,分别移取水样2.0 mL,按照本方法进行测定,同时进行加标回收实验,取水样2.0 mL加入 7.88×10-5mol/L H2O2标准工作液 0.3 mL,测定结果见表3。

表3 水样的分析结果及回收率(n=5)Table 3 Analytical results and recovery of samples(n=5)
3 结 论
本文首次将H2O2-Mn2+的类Fenton反应和纳米银-Mn2+的络合反应相结合,建立了一种测定痕量H2O2的共振散射光谱新方法,检测H2O2的线性范围为7.88×10-8~1.42×10-5mol/L,检出限为4.04×10-8mol/L,方法灵敏度较高,线性范围较宽。本文同时对体系的反应机理进行了探讨,并阐述了引起纳米银聚集状态以及导致共振散射强度发生变化的原因。
[1] Veal E A,Day A M,Morgan B A.Mol.Cell,2007,26(1):1-14.
[2] Sang Y,Zhang L,Li Y F,Chen L Q,Xu J L,Huang C Z.Anal.Chim.Acta,2010,659:224-228.
[3] Hirsch I,Prell E,Weiwad M.Anal.Biochem.,2014,456:22-24.
[4] Wang Y,Sun M,Wang L X.Phys.Test.Chem.Anal.:Chem.Anal.(王瑜,孙觅,王立霞.理化检验:化学分册),2013,49(1):91-93.
[5] Nagaraja P,Prakash J S,Asha S C,Bhaskara B L, Kumar S A.Environ.Monit.Assess.,2012,184(10):5983-5988.
[6] Huang J,Chang Q,Jiang G D,Qiu Y,Tang H Q.Phys.Test.Chem.Anal.:Chem.Anal.(黄佳,常青,江国栋,邱阳,唐和清.理化检验:化学分册),2014,50(4):417-420.
[7] Zhou T Y,Yao Q H,Zhao T T,Chen X.Talanta,2015,141:80-85.
[8] Huang H,Xu M,Gao Y,Wang G N,Su X G.Talanta,2011,86:164-169.
[9] Yang X D,Zhao P L,Qu J Q,Liu R Y.Luminescence,2014,30:592-599.
[10]Hu H C,Jin H J,Chai X S.J.Chromatogr.A,2012,1235:182-184.
[11]Sun J,Mao Y,Li G,Zhang L.J.FoodSaf.Qual.(孙佳,毛燕,李刚,张丽.食品安全质量检测学报),2013,4(6):1873-1879.
[12]Wei J J,Hu Y,Peng X S,Zhu L L,Du J Y.J.Instrum.Anal.(尉洁净,胡越,彭心声,朱丽丽,杜江燕.分析测试学报),2015,34(4):401-406.
[13]Kong F Y,Li W W,Wang J Y,Fang H L,Fan D H,Wang W.Anal.Chim.Acta,2015,884:37-43.
[14]Yao H,Li X H,Liu H.J.Instrum.Anal.(姚慧,李小红,刘欢.分析测试学报),2014,33(1):33-38.
[15]Yang S L,Li G,Wang G F,Zhao J H,Hu M F,Qu L B.Sens.ActuatorsB,2015,208:593-599.
[16]Wei X P,Liu T,Qu T Y,Li J P.J.Instrum.Anal.(魏小平,刘涛,屈太原,李建平.分析测试学报),2012,31(3):332-336.
[17]Tang N L,Shan Y Q,Zhang R H,Meng X L.Anal.Methods,2015,7:8750-8756.
[18]Qin W L,Fang Y Y,Shan Y Q,Tang N L.Chin.J.Anal.Lab.(覃温露,方业毅,单雅琦,唐宁莉.分析试验室),2013,32(11):25-27.
[19]Liang A H,Jiang Z L,Tao H L.Spectrosc.SpectralAnal.(梁爱惠,蒋治良,陶慧林.光谱学与光谱分析),2007,27(1):120-122.
[20]Zhang Y,Zhang Y J,Xia X D,Hou X Q,Feng C T,Wang J X,Deng L.Chin.Chem.Lett.,2013,24:1053-1058.
[21]Han T H,Khan M M,Lee J,Cho M H.J.Ind.Eng.Chem.,2014,20:2003-2009.
[22]Song Y J,Wei W L,Qu X G.Adv.Mater.,2011,23(37):4215-4236.
[23]Peng H I,Miller B L.Analyst,2011,136(3):436-447.
[24]Farhadi K,Forough M,Molaei R,Hajizadeh S,Rafipour A.Sens.ActuatorB,2012,161(1):880-885.
[25]Ju Y,Wang X L,Li B X.J.ShaanxiNorm.Univ.:Nat.Sci.Ed.(巨芸,王雪丽,李保新.陕西师范大学学报:自然科学版),2011,39(4):51-54.
[26]Wang Z H,Shen L.J.HangzhouTeachersColl.:Nat.Sci.Ed.(王志华,沈良.杭州师范学院学报:自然科学版),2006,5(6):465-481.
[27]Zhou Y,Zhao H,Li C,He P,Peng W B,Yuan L F,Zeng L X,He Y J.Talanta,2012,97:331-335.
[28]Wang G L,Zhu X Y,Jiao H J,Dong Y M,Wu X M,Li Z J.Anal.Chim.Acta,2012,747:92-98.
[29]Kasprzak K S.FreeRadicalBio.Med.,2002,32(10):958-967.
[30]Yu H D,Fang R,Chen S M,Zou G L.ActaChim.Sin.(于怀东,方茹,陈士明,邹国林.化学学报),2005,63(14):1357-1360.
[31]Zhao Y M,Yang S H,Ni Y H.PaperandPaperMaking(赵雨萌,杨淑蕙,倪永浩.纸和造纸),2002,6:55-56.
[32]Liang A H,Zhang N N,Jiang Z L,Liu R J.Sci.ChinaB(梁爱惠,张南南,蒋治良,刘荣进.中国科学 B辑:化学),2008,38(1):35-42.
Determination of Trace Hydrogen Peroxide by Resonance Light Scattering Method Based on Mn(Ⅱ)-Silver Nanoparticles System
TANG Ning-li*,CHEN Yong-ning,ZHANG Rong-hui,QIN Wen-lu
(College of Chemistry and Biological Engineering,Guilin University of Technology,Guilin541004,China)
According to the system resonance light scattering intensity will change with the variation of AgNPs aggregation state before and after the reaction,a novel resonance light scattering(RLS) method for the quantitative determination of trace hydrogen peroxide was developed using silver nanoparticles(AgNPs) as the resonance scattering probe.The silver nanoparticle with an average particle size of 4 nm was synthesized through the reduction of silver nitrate by sodium borohydride,using trisodiumcitrate as the protecting agent.The detection of H2O2mechanism was based on that Mn2+was chelated by the citrate covering on the surface of the silver nanoparticles,inducing the aggregation of AgNPs with increasing of resonance scattering intensity of the system.When H2O2was added to the system,Mn2+participated in Fenton-like reaction with H2O2producing Mn3+and hydroxyl radical(·OH).The complexation reaction of citrate on the surface of the AgNPs with Mn3+formed unstable complexes,and ·OH oxidatively etched AgNPs.Both of the two reactions could result in non-aggregation of AgNPs as well as relatively weakening resonance scattering intensity of the system.Under the optimal conditions,the resonance light scattering intensity of system at 411 nm was decreased linearly with concentration of H2O2over the range of 7.88×10-8-1.418×10-5mol/L,with a correlation coefficient of 0.996 7 and a detection limit of 4.04×10-8mol/L.This method was applied in the analysis of hydrogen peroxide in the lake and rain water samples with satisfactory results.
hydrogen peroxide;silver nanoparticles;resonance light scattering;Fenton-like reaction
2016-01-14;
2016-03-09
国家自然科学基金(21165008)
唐宁莉,硕士,教授,研究方向:分子光谱分析,Tel:0773-2538431,E-mail:ysshiyanshi@163.com
10.3969/j.issn.1004-4957.2016.09.003
O657.3;TQ123.6
A
1004-4957(2016)09-1094-06
猜你喜欢
江苏调味副食品(2021年1期)2021-04-01
化学与粘合(2020年6期)2020-03-08
中国洗涤用品工业(2019年4期)2019-05-11
科技视界(2017年25期)2017-12-11
中华老年口腔医学杂志(2016年1期)2017-01-15
河北医学(2016年5期)2016-12-01
应用化工(2015年2期)2015-07-13
应用化工(2014年1期)2014-08-16
长江大学学报(自科版)(2014年4期)2014-03-20
无机化学学报(2014年5期)2014-02-28
