
改性双基推进剂中化学安定剂作用机制的理论研究
2016-11-03 01:14唐秋凡樊学忠李吉祯付小龙毕福强
固体火箭技术 2016年1期
唐秋凡,樊学忠,李吉祯,付小龙,毕福强
(西安近代化学研究所,西安 710065)
改性双基推进剂中化学安定剂作用机制的理论研究
唐秋凡,樊学忠,李吉祯,付小龙,毕福强
(西安近代化学研究所,西安710065)
利用Gaussian 98计算软件,模拟了不同化学安定剂的安定机理,分析比较了二苯胺(DPA)、二苯脲(DPU)、N-甲基对硝基苯胺(MNA)吸收氮氧化物的反应活化能,以及它们在不同温度下热力学和动力学参数的变化。结果表明,标准温度下,3种化学安定剂安定作用由强到弱依次为MNA>DPA>DPU,且温度升高会抑制DPA对氮氧化物的吸收。因此,高温条件下,MNA和DPU更具有热力学优势。
Gaussian 98;化学安定剂;过渡态理论;活化能
0 引言
改性双基固体推进剂在贮存期内,保持其化学性能不发生超过允许变化范围的能力,称之为化学安定性。常用的硝化纤维素(NC)、硝化甘油(NG)等含硝酸酯基化合物是改性双基固体推进剂中的主要能量组分,且这类硝酸酯组分很容易发生热分解,这种分解服从阿累尼乌斯关系式,理论上在任何温度下都能发生。推进剂中的硝酸酯热分解反应生成氧化氮自由基,而氧化氮自由基又可催化加速硝酸酯分解,即RONO2+H2O—ROH+H+NO3-;生成的硝酸又分解成NO2,部分NO2又与硝酸酯反应,本身变为NO,如此反复循环的自催化分解造成推进剂性能下降,且伴随着硝酸酯的分解,还会产生大量的热,会导致推进剂温度的升高,甚至发热燃烧、爆炸[1-2]。实际配方中,需要添加苯胺衍生物、酰胺类和苯酚类衍生物等化学安定剂,吸收推进剂组分分解放出的酸、氧化氮及其自由基,抑制硝酸酯的这种自催化分解,达到提高含硝酸酯类推进剂化学安定性的目的。
目前,国外对化学安定剂安定作用的研究多关注安定反应及其动力学[3-4];国内化学安定剂在推进剂的应用多以经验为主,有关理论方面的研究报道较少。丁黎等[5]采用密度泛函理论B3LYP方法和6-31g(d)基组,计算了安定剂结合H+的反应能变,以解释DPA、C2、AKⅡ 3种安定剂安定化作用的强弱;徐文[6]采用密度泛函理论B3LYP方法和6-31g(d)基组,计算DPA、C2、AKⅡ 3种安定剂与NO的结合能来研究它们的相互作用。如今在改性双基推进剂应用中遇到的许多问题,如拓宽改性双基固体推进剂贮存和使用温度、设计合成新型有效的安定剂等,缺乏有效的理论指导。因此,急需开展化学安定剂安定机理的深入研究。
本研究利用Gaussian 98[7]计算软件模拟不同化学安定剂的安定机理,分析比较化学安定剂二苯胺(DPA)、二苯脲(DPU)、N-甲基对硝基苯胺(MNA)吸收氮氧化物的反应活化能,以及它们在不同温度下热力学和动力学参数的变化,来比较不同安定剂对双基系固体推进剂的安定化强弱。
1 计算方法
以过渡态理论为依据,利用Gaussian 98计算软件模拟不同化学安定剂的安定反应中反应物、过渡态和产物的结构,并进行IRC反应路径分析,通过比较反应物、过渡态和产物三者结构和能量的变化推测反应机理。在过渡态理论[8]的基础上,利用Chem3D、GaussView程序,搭建可能存在的各个反应物、过渡态、中间体和产物的构型,经过分析比较文献[5-6]中的计算方法,发现半经验方法虽然所用时间短耗时少,但所得结果与实验值相比误差较大,而B3LYP方法充分考虑了电子相关性,其所得计算结果相比于比半经验方法、HF方法要更准确,且和HF方法耗时相当,甚至更少。综合时间和计算精度的问题,本文采用B3LYP方法和6-31g(d)基组。
为了研究反应的动力学性质以及研究温度对反应的影响,在B3LYP/6-31g(d)理论水平上,计算出不同温度下反应热力学函数的改变值ΔH、ΔS和ΔG,利用过渡态理论,通过Eyring方程式(1)计算相关动力学参数,以比较不同安定剂在改性双基固体推进剂中的
安定化作用强弱。
(1)
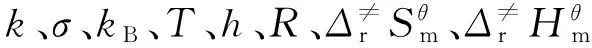
2 结果与讨论
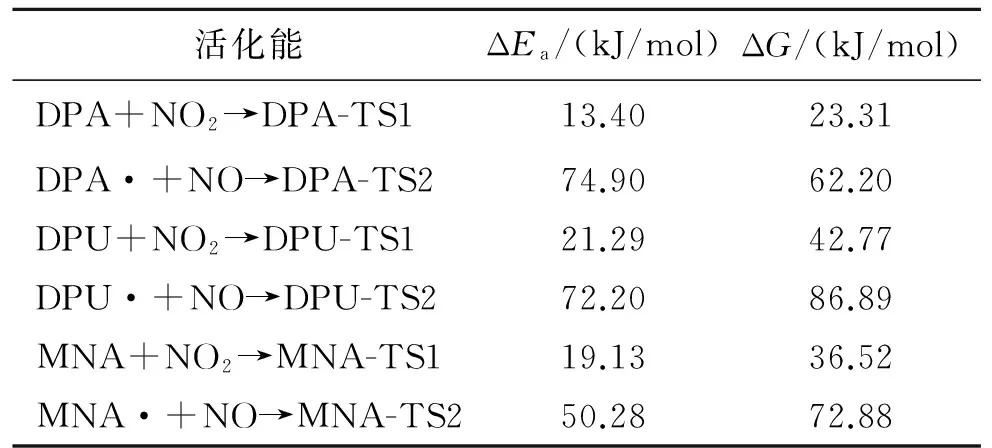
有研究表明[9-10],DPA不能直接与NO反应,而能与NO2反应。Lussier等[11]利用HPLC,对和不同体积NO2反应的DPA的产物进行分析。通过对反应产物中各个组分含量变化的比较,他们提出DPA安定反应机理见图1,本文在此机理的基础上进行计算模拟。
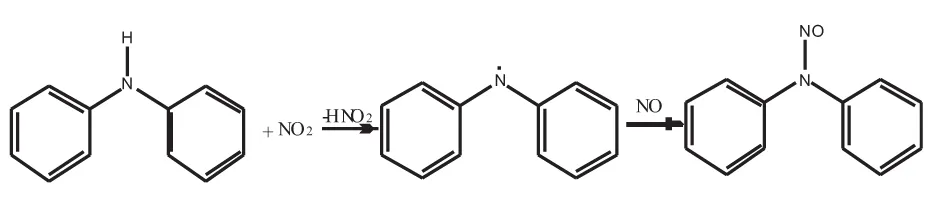
图1 DPA安定反应机理
2.1过渡态结构及其IRC反应路径分析
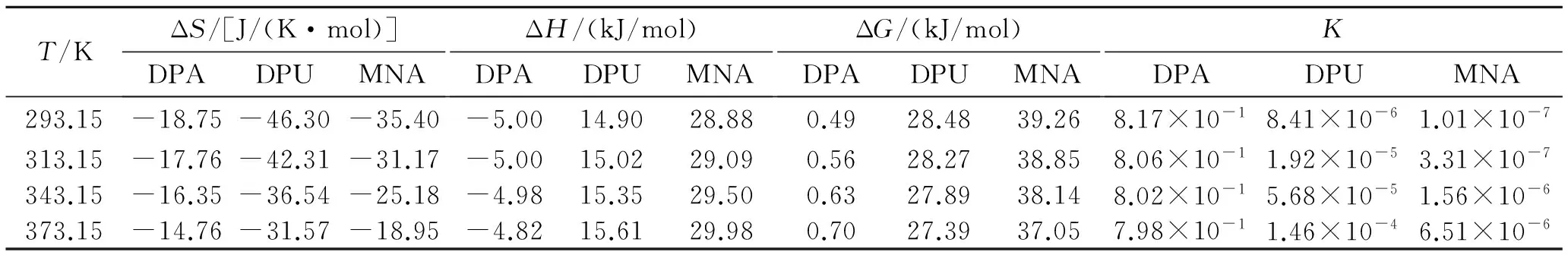
计算采用Gaussian 98程序,采用密度泛函理论(DFT)中的B3LYP方法,在6-31g(d)基组水平上计算了DPA安定反应中的反应物、过渡态、中间体和产物结构。每个过渡态经IRC反应路径分析后,对抛物线两端能量最低的2个结构进行优化。过渡态以及优化后的反应物、中间体和产物的络合结构构型见图2,反应过程中变化的主要几何构型参数列于表1。表1中,R表示2个原子之间键长;A表示3个原子形成的键角;S表示4个原子形成的二面角。
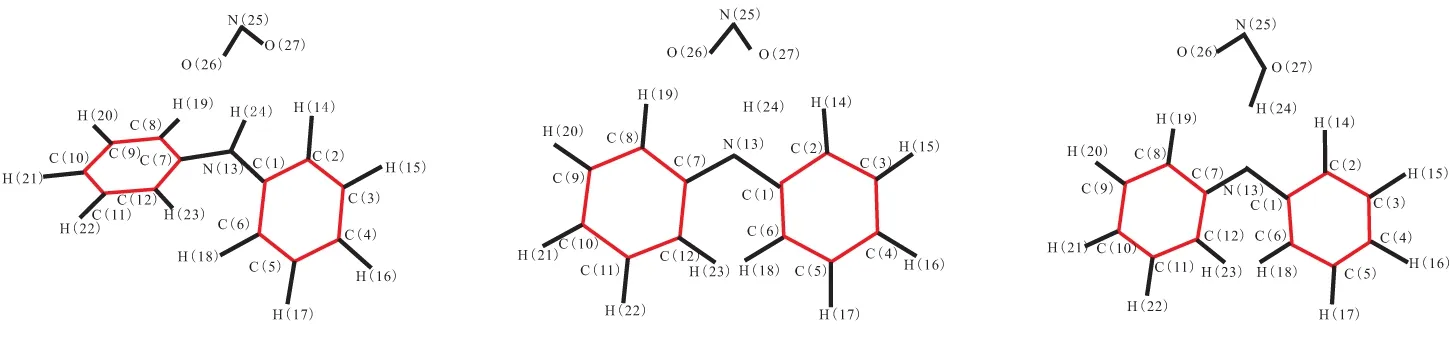
(a)DPA+NO2 (b)DPA-TS1 (c)DPA·+HNO2
(d)DPA·+NO (e)DPA-TS2 (f)N-NO-DPA

构型参数DPA+NO2DPA-TS1DPA·+HNO2构型参数DPA·+NODPA-TS2N-NO-DPAR(1,13)0.13970.13700.1371R(1,13)0.13680.14250.1426R(7,13)0.13980.13850.1375R(7,13)0.13690.14350.1435R(13,24)0.10160.11990.1806R(13,24)0.28520.15890.1373R(24,27)0.20710.13350.1019R(24,25)0.11610.11760.1218R(25,26)0.12110.12300.1202A(1,13,7)123.31118.98121.99R(25,27)0.12230.12990.1361A(13,24,25)102.75111.16115.39A(1,13,7)129.26126.65123.34D(7,13,24,25)83.16125.04118.27A(24,27,25)102.47111.12110.10————A(26,25,27)128.06115.43114.36————D(13,24,27,25)-164.27-125.40-164.27————D(24,27,25,26)-3.02-9.11-3.02————
在DPA吸收氮氧化物的反应过程中,首先NO2夺取N(13)上的H(24),经过过渡态(DPA-TS1),形成中间体(DPA·+HNO2)。在此过程中,N(13)—H(24)之间的键长不断增大,从最开始反应物的0.101 6 nm,到过渡态的0.119 9 nm,最后到中间体0.180 6 nm。可见,H(24)在逐渐脱离DPA上的氨基N(13)。同时,随着H(24)的离去,C(1)—N(13)和C(7)—N(13)之间的键长及C(1)—N(13)—C(7)间的键角也随之发生了变化,键长从原来的0.139 7 nm和0.139 8 nm减小到0.137 1 nm和0.137 5 nm,键角从原来的129.26°减小到123.34°。H(24)和NO2中O(27)的键长在不断减小,从不成键的0.201 7 nm,到过渡态的0.133 5 nm,最后到形成O(27)—H(24)单键的0.101 9 nm。而且随着H(24)的不断靠近,NO2中的2个氮氧键也随之发生变化:反应前后N(25)—O(26)的键长分别为0.121 1 nm和0.120 2 nm,略有减小,但N(25)—O(27)的键长从原来的0.122 3 nm增大到0.136 1 nm,说明N(25)—O(27)变为单键。因此,最后形成的是HNO2和自由基DPA·。而过渡态中,H(24)处于N(12)和O(27)之间,他们的键长分别为0.119 9 nm和0.133 5 nm,H(24)处于一种既不与N(12)成键也不与O(27)成键的中间状态。
第一步反应形成的自由基DPA·与HNO2分解产生的NO或者推进剂中硝酸酯分解产生的NO发生反应。在此过程中,N(13)—N(24)的键长不断减小,由开始的0.285 2 nm,到过渡态的0.158 9 nm,到产物的0.137 3 nm,可见NO在不断地靠近自由基DPA·,并形成N(13)—N(24)单键。随着NO的不断靠近,DPA·中C(1)—N(13)和C(7)—N(13)间的键长及C(1)—N(13)—C(7)间的键角也随之发生了变化,键长从原来的0.136 8 nm和0.136 9 nm增大到0.142 6 nm和0.143 5 nm,键角从原来的123.31°减小到121.99°。而N(24)—O(25)间的键长从开始的0.116 1 nm,增加到过渡态(DPA-TS2)的0.117 6 nm,最后到N(13)—N(24)成键后的0.121 8 nm。
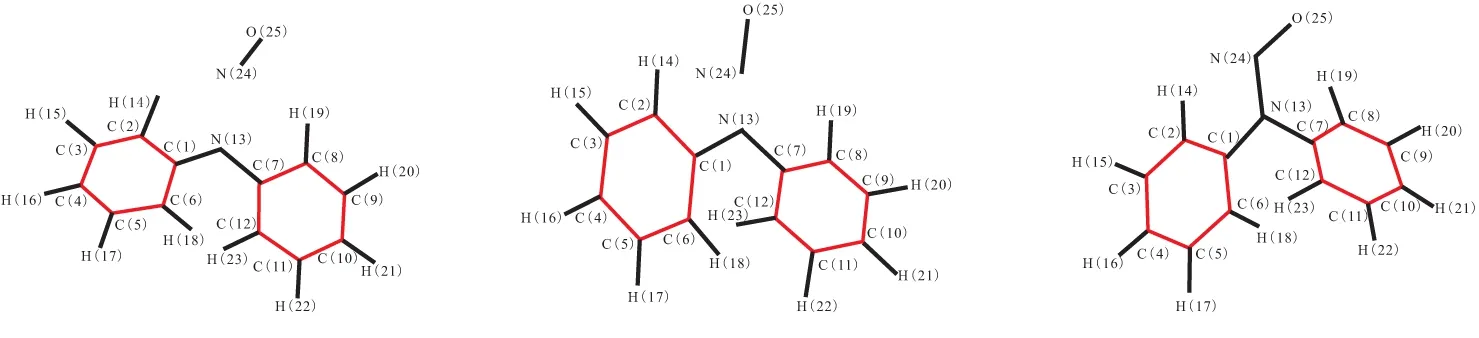
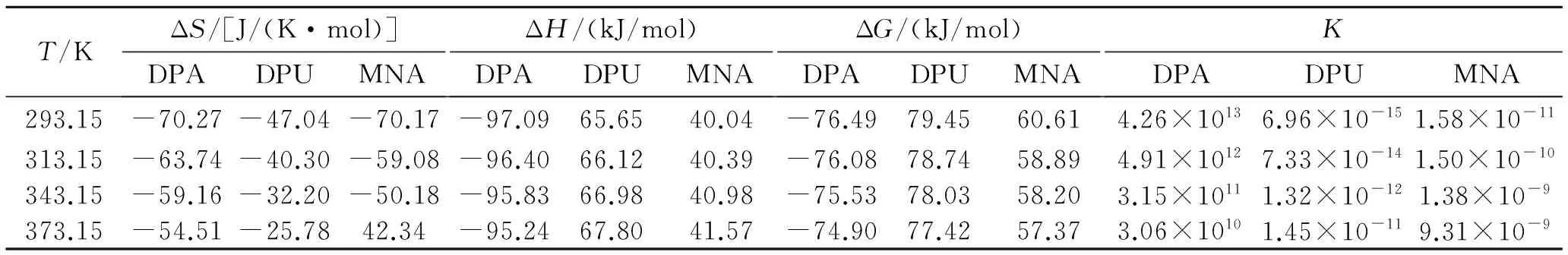
对二苯脲(DPU)和N-甲基对硝基苯胺(MNA)这类结构和DPA类似,有相似反应机理的物质,利用Gaussian 09程序,采用密度泛函理论(DFT)中的B3LYP方法,在6-31g(d)基组水平上进行相同的计算。DPU反应过渡态、反应物、中间体和产物的络合结构构型见图3,反应过程中变化的主要几何构型参数列于表2。MNA反应过渡态、反应物、中间体和产物的络合结构构型见图4,反应过程中变化的主要几何构型参数列于表3。
在DPU和MNA吸收氮氧化物的反应过程中,键长、键角和二面角的变化规律和DPA的相同。首先是安定剂中的氨基氮上的N—H键长不断增加,而H与NO2中的一个O的距离不断减小。H从开始的与氨基N成键状态到脱离N,且与NO2中的O形成O—H单键。而NO2中的N—O键长,一个略有减小,另一个明显增大,形成N—O单键,说明在第一阶段反应最后形成的产物是安定剂失去氨基氮上的H后形成的自由基和HNO2。之后,第一步反应形成的自由基与HNO2分解产生的NO或者推进剂中硝酸酯分解产生的NO发生反应,安定剂氨基氮与NO的N—N键长不断减小,最后成键,且NO的N—O键长在不断增加。最后,N—O的键长处于亚硝基的键长范围,说明此过程是NO在不断靠近,且与自由基结合形成亚硝基产物。
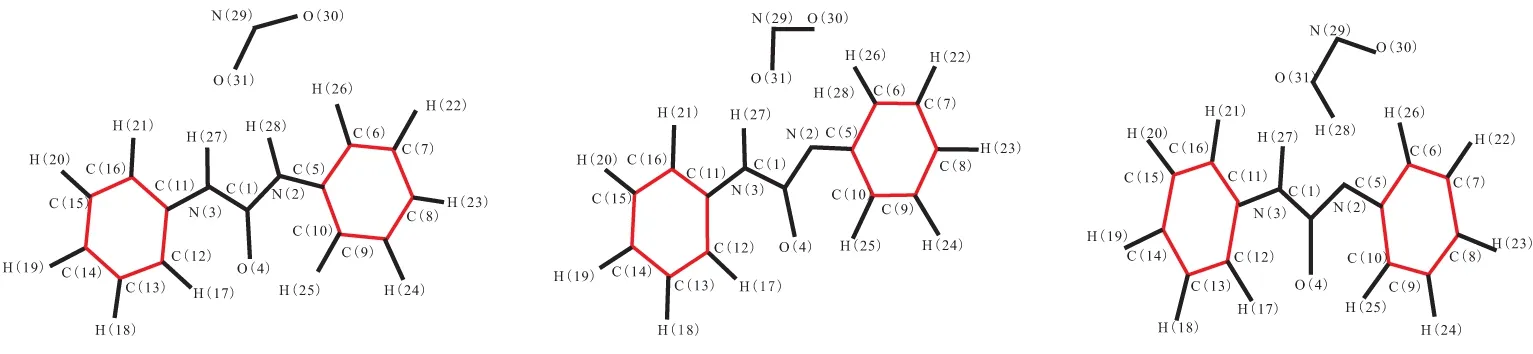
(a)DPU+NO2 (b)DPU-TS1 (c)DPU·+HNO2

(d)DPU·+NO (e)DPU-TS2 (f)N-NO-DPU
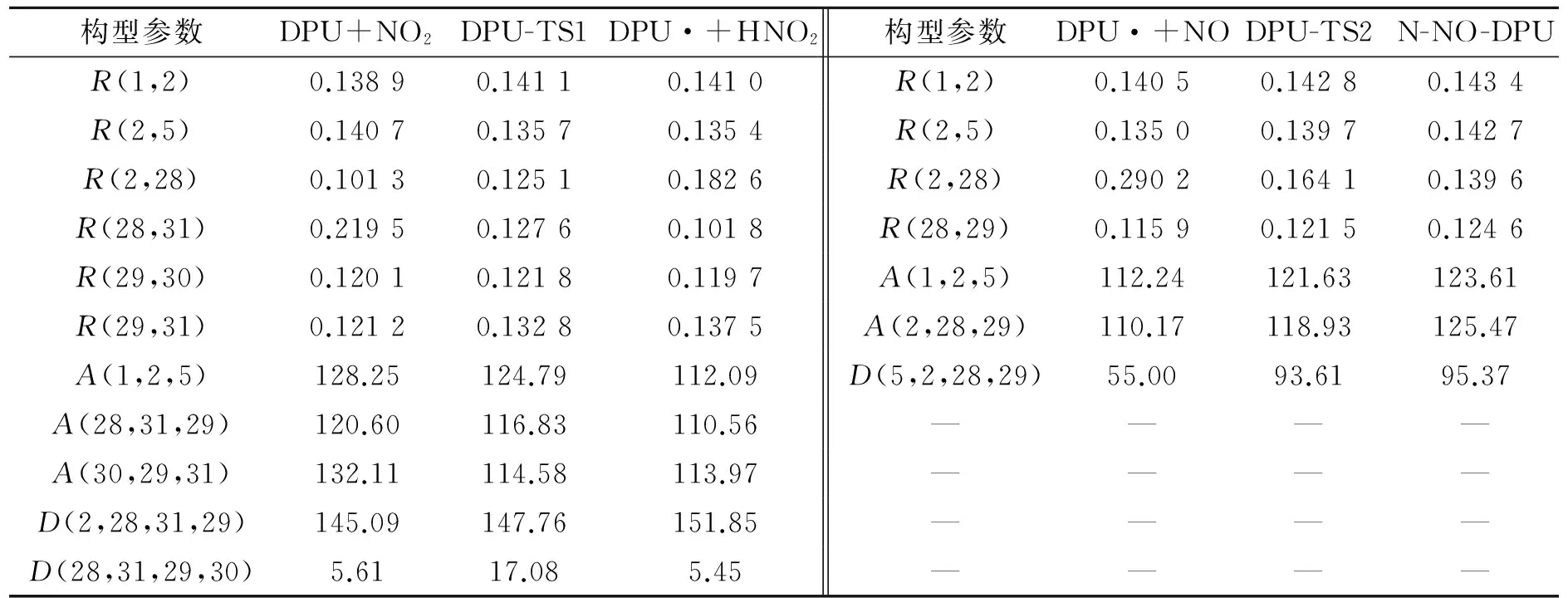
构型参数DPU+NO2DPU-TS1DPU·+HNO2构型参数DPU·+NODPU-TS2N-NO-DPUR(1,2)0.13890.14110.1410R(1,2)0.14050.14280.1434R(2,5)0.14070.13570.1354R(2,5)0.13500.13970.1427R(2,28)0.10130.12510.1826R(2,28)0.29020.16410.1396R(28,31)0.21950.12760.1018R(28,29)0.11590.12150.1246R(29,30)0.12010.12180.1197A(1,2,5)112.24121.63123.61R(29,31)0.12120.13280.1375A(2,28,29)110.17118.93125.47A(1,2,5)128.25124.79112.09D(5,2,28,29)55.0093.6195.37A(28,31,29)120.60116.83110.56————A(30,29,31)132.11114.58113.97————D(2,28,31,29)145.09147.76151.85————D(28,31,29,30)5.6117.085.45————

(a)MNA+NO2 (b)MNA-TS1 (c)MNA·+HNO2

(d)MNA·+NO (e)MNA-TS2 (f)N-NO-MNA
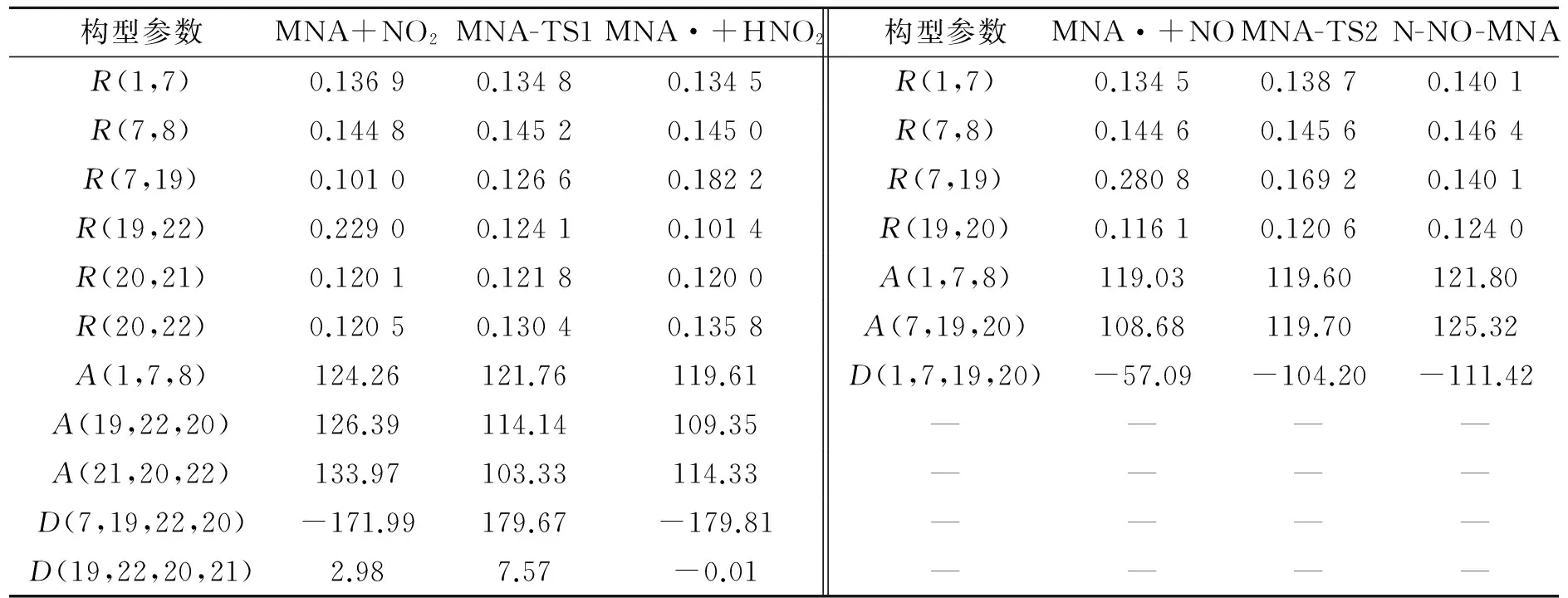
构型参数MNA+NO2MNA-TS1MNA·+HNO2构型参数MNA·+NOMNA-TS2N-NO-MNAR(1,7)0.13690.13480.1345R(1,7)0.13450.13870.1401R(7,8)0.14480.14520.1450R(7,8)0.14460.14560.1464R(7,19)0.10100.12660.1822R(7,19)0.28080.16920.1401R(19,22)0.22900.12410.1014R(19,20)0.11610.12060.1240R(20,21)0.12010.12180.1200A(1,7,8)119.03119.60121.80R(20,22)0.12050.13040.1358A(7,19,20)108.68119.70125.32A(1,7,8)124.26121.76119.61D(1,7,19,20)-57.09-104.20-111.42A(19,22,20)126.39114.14109.35————A(21,20,22)133.97103.33114.33————D(7,19,22,20)-171.99179.67-179.81————D(19,22,20,21)2.987.57-0.01————
综上所述,几种安定剂的安定机理:(1)NO2夺取安定剂氨基N上的H形成自由基和HNO2;(2)HNO2分解产生NO和NO2气体;(3)由硝酸酯组分分解的及HNO2分解的NO与自由基结合生成亚硝基产物。
2.2能量变化及活化能分析
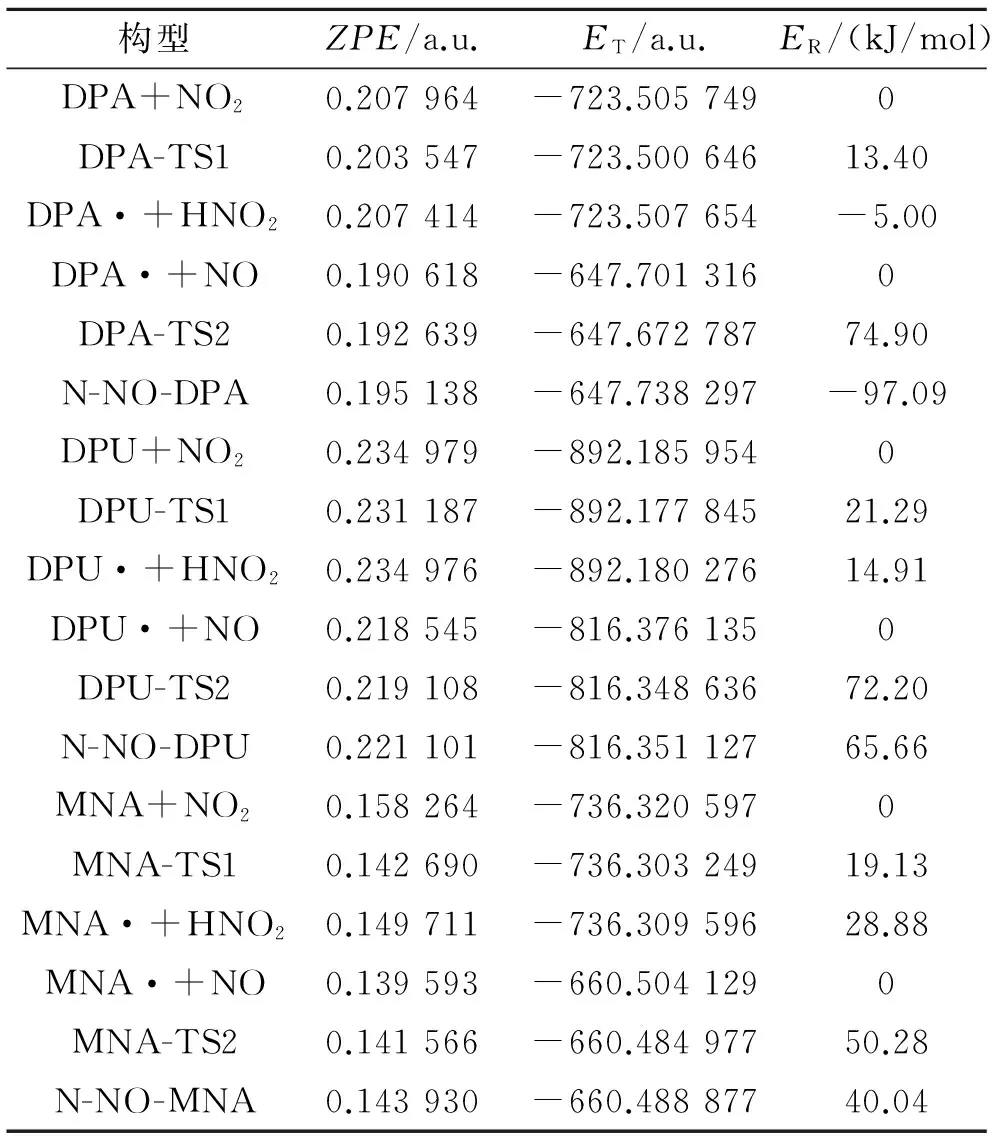
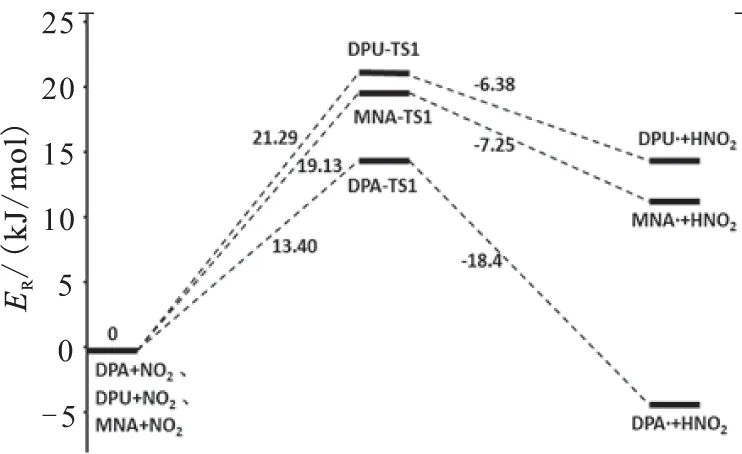
为了计算各个反应的能量变化,表4列出了各个构型的能量信息,ZEP为零点能,ET为总能量,ER为相对能量,得到了2个不同阶段反应的能级图,见图5。表5列出了反应物到中间体、中间体到产物的能量变化,表6列出了各个反应的活化能。
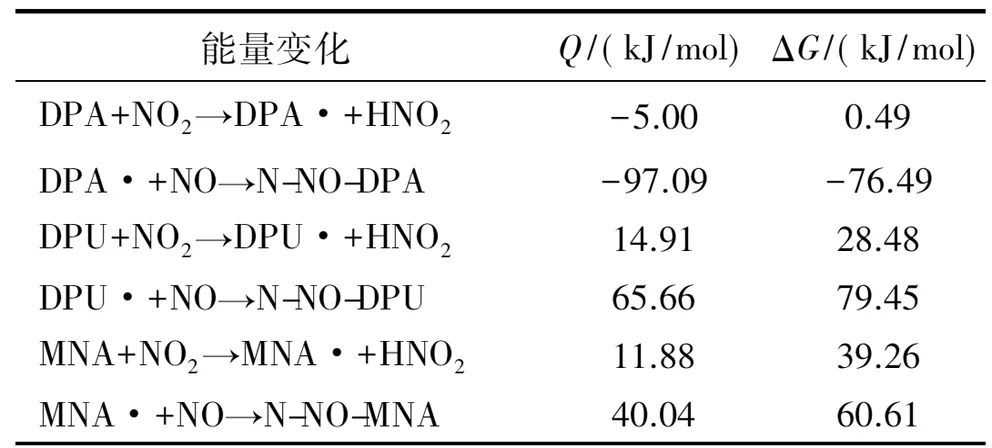
由表5的产物、中间体和产物的能量数据可知,DPA第一阶段反应的Q和ΔG分别是-5.00 kJ/mol和0.49 kJ/mol,第二阶段反应分别为-97.09 kJ/mol和-76.49 kJ/mol,说明这2个反应的过程是放热的。而DPU和MNA的Q和ΔG都是大于零,且DPU 2个反应的Q和ΔG都大于MNA,说明反应是吸热的,吸收热量的大小为DPU>MNA。
由表6列出的各个反应的活化能数据可知,DPA、DPU、MNA 3种安定剂到过渡态1(TS1)反应活化能ΔEa分别为13.40、21.29、19.13 kJ/mol,吉布斯自由能变ΔG分别是23.31、42.77、36.52 kJ/mol。可见,活化能及吉布斯自由能变大小顺序一致,为DPA 综合上述两表数据,可得到以下结论:在标准温度下,对于整个反应,MNA吸收氮氧化物的活化能和吉布斯自由能变是3种化学安定剂中最小的,3种化学安定剂安定顺序为MNA>DPA>DPU。 2.3热力学和动力学分析 在B3LYP/6-31G理论水平上,分别计算出反应热力学函数改变值ΔH、ΔS和ΔG。 (2) (3) 表4 所用结构构型的能量 根据式(2)和式(3)计算出了反应的平衡常数K,以及表观速率常数k随温度的变化值,平衡常数K数据见表7和表8,表观速率常数k随温度的变化数据见表9和表10。 (a)第1阶段 (b)第2阶段 能量变化Q/(kJ/mol)ΔG/(kJ/mol)DPA+NO2→DPA·+HNO2-5.000.49DPA·+NO→N-NO-DPA-97.09-76.49DPU+NO2→DPU·+HNO214.9128.48DPU·+NO→N-NO-DPU65.6679.45MNA+NO2→MNA·+HNO211.8839.26MNA·+NO→N-NO-MNA40.0460.61 表6 各个反应的活化能 表7 第1阶段反应热力学参数 表8 第2阶段反应热力学参数 表9 动力学参数和 表10 动力学参数(A和k) 从表7数据可知,在20~100 ℃范围内,3种安定剂第1阶段反应的ΔS均小于零。DPA的ΔH小于零,ΔG大于零,而且均有逐渐增加的趋势,平衡常数K随温度升高而减小,说明DPA第1阶段是一个熵减小、放热的反应,温度的升高,抑制了DPA第1阶段的反应。另外,2种安定剂的ΔH和ΔG均大于零,且随温度升高而逐渐减小,平衡常数K随温度升高而增大,说明DPU和MNA第1阶段是一个熵减小、吸热的反应,高温条件有利于DPU和MNA第1阶段的反应。 从表8数据可知,在20~100 ℃范围内,3种安定剂第2阶段反应的ΔS均小于零。DPA的ΔH和ΔG大于零,且均有逐渐增加的趋势,平衡常数K随温度升高而减小,说明DPA第2阶段也是一个熵减小、放热的反应,温度的升高,同样会抑制了DPA第2阶段的反应。另外,2种安定剂的ΔH和ΔG均大于零,且随温度升高而逐渐减小,平衡常数K随温度升高而增大,说明DPU和MNA第2阶段也是一个熵减小、吸热的反应,高温条件有利于DPU和MNA第2阶段的反应。 从表9和表10的数据可知,在20~100 ℃温度范围内,随着温度的升高,3种化学安定剂第1阶段反应速率常数均增大,第2阶段DPA反应速率常数减小,DPU和MNA反应速率常数增大。说明这个温度范围内,高温条件有利于DPU和MNA吸收氮氧化物反应的进行。由此可得出结论,高温条件有利于DPU和MNA对氮氧化物的吸收。而对DPA来说,随着温度的升高,反而抑制了其对氮氧化物的吸收。高温条件下,DPU和MNA更具有热力学优势。 通过Gaussian 98计算软件,模拟了二苯胺(DPA)、二苯脲(DPU)、N-甲基对硝基苯胺(MNA)3种化学安定剂吸收氮氧化物的反应机理:NO2夺取安定剂氨基N上的H,形成自由基和HNO2,HNO2分解产生NO和NO2气体,由硝酸酯组分分解的以及HNO2分解的NO与自由基结合生成亚硝基产物。理论研究表明:标准温度下,3种化学安定剂安定作用由强到弱依次为MNA>DPA>DPU,随着温度的升高,从吉布斯自由能和平衡常数的变化来看,DPA吸收氮氧化物的反应受到抑制,而DPU和MNA则相反。因此,在20~100 ℃温度范围内,MNA的效果是3种化学安定剂中最好的。 [1]Bohn M A.Prediction of life times of propellants-improved kinetic description of stabilizer consumption[J].Propellants,Explosives,Pyrotechnics,1994,19(5):266-269. [2]Yim Joo-yin.Stabilizer consumption by accelerated aging of PEG/RDX propellant[C]//29th ICT.Karlsruhe,1998:45.1-45.9. [3]Anton Chin,Daniel S Ellison.Mechanistic approach to study the moisture and acidity effect on the stability of single and double based propellants[C]//28th International Pyrotechnics Seminar.2001:173-186. [4]Lussier Louis Simon,Eric Bergeron,Hélène Gagnon.A mothed to characterize gun power stabilizers[C]//37th International Annual Conference of ICT.2006. [5]丁黎,郑朝民,翟高红,等.推进剂安定剂与硝酸酯(NG-NC)相互作用研究[J].固体火箭技术,2014,37(4):252-529. [6]徐文.RDX和多元硝酸酯的热解及AKⅡ、C2水解和安定机理的理论研究[D].西安:西北大学,2011. [7]Frisch M J,Trucks G W,Schlegel H B,et al.Gaussian 98(Revision A.11)[CP/CD].Gaussian:Inc.of Pittsburgh PA,2001. [8]Laidler K J,King M C.Development of transition-state theory[J].The Journal of Physical Chemistry,1983,87(15):2657-2664. [9]Alm A.Studies on reactions between nitrogen dioxide and diphenylamine compounds[C]//J.Hansson(ed),1st Symp.On Chemical Problem Connected with the Stability of Explosives.Stockholm,1968:162-178. [10]Isler J.Etude du pouvoir stabilisant de la diphenylamine et de ses principaux derives[R].Note Technique No.77/83 GER Py,Groupe d'Etudes et Recherches en Pyrotechnie,Direction des Construction et Armes Navales de Toulon,France(1983). [11]Lussier L S,Gagnon H,Bohn M A.On the chemical reactions of diphenylamine and its deriv atives with nitrogen dioxide at normal storage temperature conditions[J].Propellants,Explosives,Pyrotechnics,2000,25(3):117-125. (编辑:刘红利) Theoretical research on mechanism of chemical stabilizers in composite modified double base propellant TANG Qiu-fan,FAN Xue-zhong,LI Ji-zhen,FU Xiao-ong,BI Fu-qiang (Xi'an Modern Chemistry Research Institute,Xi'an710065,China) The reaction activation energy and changes of thermodynamic and kinetic parameter of different chemical stabilizers including diphenylamine(DPA),diphenyl urea(DPU) and P-nitro-N-methylamine (MNA) at different temperatures were analyzed by using Gaussian 98 to simulate their stability mechanism.The result indicate that the influence on stability under the normal temperature of different stabilizers from strong to weak is MNA>DPA>DPU,and the DPA on absorption of nitrogen oxides would be inhibited with the increase of temperature,so MNA and DPU have more thermodynamic advantages under higher temperature. Gaussian 98;chemical stabilizer;transition-state theory;activation energy 2015-04-13; 2015-06-08。 唐秋凡(1990—),男,硕士生,从事固体推进剂研究。E-mail:asdasd953@163.com V512 A 1006-2793(2016)01-0065-08 10.7673/j.issn.1006-2793.2016.01.012
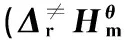

3 结论
猜你喜欢
四川大学学报(自然科学版)(2022年4期)2022-07-22
北京航空航天大学学报(2022年5期)2022-06-06
原子与分子物理学报(2022年3期)2022-03-05
中学生数理化·高一版(2020年11期)2020-12-14
青岛大学学报(工程技术版)(2019年2期)2019-09-10
中学化学(2019年2期)2019-07-08
电脑知识与技术(2018年3期)2018-03-21
哈尔滨理工大学学报(2017年1期)2017-04-08
科技视界(2016年24期)2016-10-11
中学化学(2015年8期)2015-12-29
