
桥联茂钛配合物催化丙烯聚合反应选择性的理论研究
2016-11-02 02:12唐艳辉王志栋
高等学校化学学报 2016年10期
唐艳辉, 王志栋
(1. 北京服装学院材料科学与工程学院, 北京 100029;2. 北京化工大学理学院, 化工资源有效利用国家重点实验室, 北京 100029)
桥联茂钛配合物催化丙烯聚合反应选择性的理论研究
唐艳辉1, 王志栋2
(1. 北京服装学院材料科学与工程学院, 北京 100029;2. 北京化工大学理学院, 化工资源有效利用国家重点实验室, 北京 100029)
采用密度泛函方法对3种不同类型的硅桥联茂钛配合物[Me2SiN(Me4Cp)TiCl2(A), Me2SiCpFluTiCl2(B)及Me2SiInd2TiCl2(C)]催化丙烯聚合反应的选择性进行了理论研究. 计算结果表明, 硅桥联茂金属配体的空间结构是其催化烯烃聚合反应的区域选择性和立体选择性的主要原因. 聚合过程中,α-烯烃配位有1,2插入(一级插入)和2,1插入(二级插入)2种方式, 3种硅桥联茂金属催化剂均表现为烯烃的一级插入, 这种区域选择性与催化剂硅桥联配体的刚性结构密切相关. 对烯烃聚合反应链增长机理进行了理论计算, 结果表明, 具有Cs对称性的Me2SiN(Me4Cp)TiCl2和Me2SiCpFluTiCl2催化丙烯聚合分别得到无规立构和间规立构的聚烯烃产物, 而具有C2对称性的Me2SiInd2TiCl2催化丙烯聚合得到等规立构的聚烯烃产物, 与实验结果一致.
硅桥联茂金属配合物; 烯烃聚合反应; 反应选择性; 反应机理; 理论研究
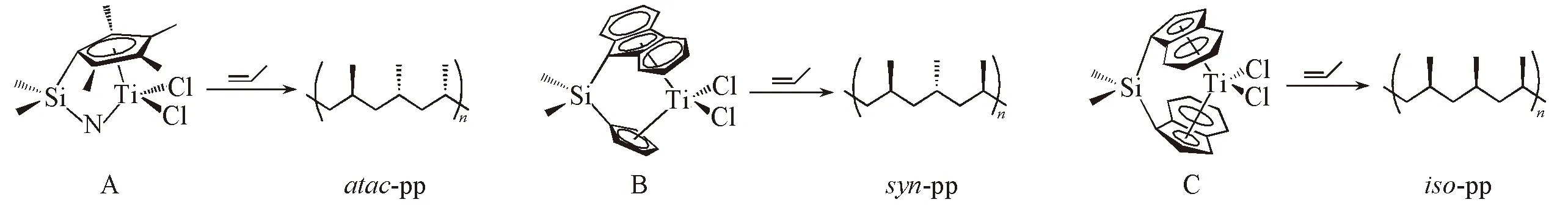
Scheme 1 Three types of bridged-metallocene catalysts and their polypropylene products
高分子聚烯烃在日常生活中发挥着重要的作用, 而聚烯烃工业的迅速发展与聚烯烃催化剂的发展密切相关[1~3]. 与传统的Ziegler-Natta催化剂相比, 单活性中心茂金属催化剂催化得到的聚合物分子量较大, 分子量分布较窄, 性能优异, 这一独特的性质使其在聚合物品种的开发上显示出了明显的优势. 用Ziegler-Natta催化剂很难实现的聚烯烃树脂的功能化, 在茂金属催化剂作用下得到了解决, 而且茂金属催化剂催化制得的聚烯烃的性能是传统催化剂催化产品难以达到的, 特别是一些高立构规整度产品的研制. 茂金属烯烃聚合催化剂催化烯烃聚合反应是当前聚烯烃催化剂领域的一个研究热点[4~9]. 茂金属配合物是一类至少包含1个环戊二烯基(Cp)或者取代的环戊二烯基配体的金属配合物. 根据茂基数量和空间结构, 茂金属烯烃聚合催化剂大致可分为4类: 非桥联双茂配合物、 桥联双茂配合物、 单茂配合物及具有限制几何结构的单茂配合物(CGC). 其中, 非桥联的茂金属催化剂多应用于乙烯均聚或与其它烯烃的共聚过程, 这类催化剂催化丙烯聚合一般会得到无规立构的聚丙烯. 由于桥基对配体的限制, 具有刚性骨架结构的桥联茂金属催化剂不仅具有高温下的结构稳定性, 而且其桥基基团或配体上的取代基的微小变化就可能对其构建的特殊空间立体效应产生巨大影响, 使其在催化合成各种立构规整性的聚合产物方面具有明显的优势[10]. 实验结果表明, 茂金属的骨架对称性对其催化性质具有很大的影响, 如CGC催化丙烯聚合的产物通常是无规立构的, 而具有Cs和C2对称性的催化剂催化的丙烯聚合产物则是间规和等规立构的[11,12], 如Scheme 1所示.
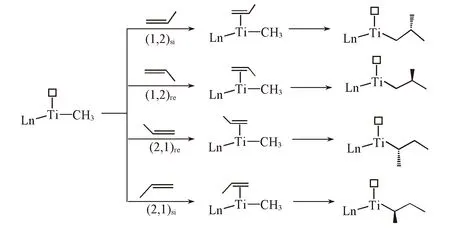
Scheme 2 Four different propylene coordination and insertion ways
茂金属催化烯烃聚合的反应机理通常采用Cossee-Arlman机理[13]. 活性中心是带正电荷的甲基配合物, 接收烯烃单体配位后, 经过四元环过渡态, 烯烃插入金属-烷基键完成链引发和链增长过程[13,14]. 对于α-烯烃聚合反应, 如Scheme 2所示, 丙烯可以(1,2)si, (1,2)re, (2,1)re和(2,1)si4种方式来完成烯烃配位插入过程. 其中(1,2), (2,1)分别代表一级插入和二级插入, re和si则分别代表丙烯以re手性面和si手性面配位. 在链增长过程中, 聚合链在不同烯烃配位点之间迁移, 然后发生烯烃单体配位插入实现链增长, 该过程不断重复直至发生链终止反应[15].
量子化学计算方法在茂金属催化剂研究领域应用广泛, Cavallo等[16]采用分子动力学方法研究了催化剂配体对配位单体的立体效应, 提出了活性中心控制模型并以此解释了一些催化现象. Olivia等[17]采用混合基组的密度泛函方法(DFT)方法并结合实验研究了茂锆金属催化剂催化苯乙烯聚合过程中电子效应对反应区位选择性的影响, 发现向苯乙烯单体引入给电子基团会使单体一级插入的趋势增大. Morokuma等[18]采用从头算分子轨道/分子力学(MO/MM)方法对硅桥联茂锆催化剂催化烯烃聚合过程进行了计算, 验证了Cossee机理并研究了催化剂的反应选择性. Cruz等[19]采用三维定量构效关系(3D-QSAR)方法研究了茂金属催化剂的结构-活性关系. 本文选择了丙烯作为聚合单体, 3种桥联钛茂催化剂(Scheme 1)作为目标催化剂, 对催化丙烯聚合链引发和链增长机理进行了计算研究, 探讨了该类催化剂催化α-烯烃聚合过程反应选择性的根源及影响因素, 为改造设计高选择性聚烯烃催化剂提供了理论参考.
1 计算方法
理论研究计算采用混合基组的DFT方法, 其中混合基组(BSI)对过渡金属中心采用基于有效核势能近似(ECP)的LANL2DZ基组, 其它原子采用6-31G(d)基组, 在ωB97XD/BSI[20]水平下优化得到了催化反应循环中的中间体、 过渡态和产物的几何构型, 并对各过渡态进行了振动分析以确认有且只有一个虚频, 同时通过反应路径分析(IRC)计算对过渡态进行进一步的验证. 所有计算均采用Gaussian 09程序[21]完成. 本文用到的计算方法在相关过渡金属配合物催化反应的理论研究中使用并表现一定的可靠性[22~29], 文中能量值如无特别指明均为自由能数据. 各驻点优化结构及能量等见图S1~图S4和表S1~表S4(见本文支持信息).

Scheme 3 Mechanism of propylene polymerization catalyzed by titanium complexes
2 结果与讨论
2.1烯烃配位插入模式
链引发步骤是整个烯烃聚合催化的起始点, 在链引发步骤中的烯烃配位插入模式是催化剂催化烯烃聚合反应产物立体规整性的根源[30]. 研究结果表明, 初始的催化剂前体并不具有催化活性, 在助催化剂作用下催化剂前体烷基化, 金属中心成为缺电子的活性中心, 催化剂的活性催化组分得以形成(Scheme 3中的1). 活性中间体1催化丙烯聚合过程中, 烯烃单体首先与金属中心配位, 生成π配合物2, 最后烯烃插入(也可称为烷基迁移过程)得到异丁基配合物中间体3.
如图1所示, 根据烯烃配位方式的不同, 烯烃配位插入过程有4条可能的反应通道, 即(1,2)si, (1,2)re, (2,1)re和(2,1)si烯烃插入模式, 其中前两者为一级插入, 后两者为二级插入.
对A, B和C 3种催化剂催化丙烯聚合反应的链引发步骤进行了计算, 各催化剂活性组分催化烯烃聚合反应路径各驻点名称以A, B和C命名(如A1代表以催化剂A的催化活性组分1). 图2给出了催化剂A催化丙烯聚合的链引发过程的能量示意图, 活性中间体A1和丙烯的能量之和为能量参考零点, 图中右侧为一级插入路径, 左侧为二级插入路径. 计算结果表明, 丙烯以一级插入的方式完成链引发的自由能垒分别为67.4和75.8 kJ/mol, 烯烃插入过程分别放热69.9和68.2 kJ/mol; 丙烯以二级插入模式的能垒分别为74.9和84.6 kJ/mol, 分别放热50.6和64.9 kJ/mol. 比较烯烃一、 二级插入路径的自由能垒, 在具有Cs对称性的催化剂A催化丙烯聚合的链引发过程中, (1,2)si路径的自由能垒较其它插入路径低, 是优势的反应路径.

Fig.2 Energy profiles of the chain initiation step of propylene polymerization catalyzed by catalyst A Values in parentheses are zero-point energies.
在链引发步骤中,α-烯烃配位插入模式的不同决定了桥联茂钛配合物中配体立体空间的区位选择性. 催化剂A的活性中间体A1的桥基和茂环构建的空间结构较对称(二面角DTi—N—Si—C2=-0.87°), 金属中心两侧可提供烯烃单体配位的空间(如图1所示). 当丙烯以(2,1)re或(2,1)si方式配位时, 丙烯的甲基靠近催化剂的桥基端, 而该区域的空间位阻较大, 配体空间口袋深且窄, 甲基与配体环境存在较强的排斥作用. 如图3所示, 4种插入模式过渡态的二面角DTi—N—Si—C2分别为7.04°, -4.87°, 10.21°和12.59°. 显然, 二级插入过渡态茂环配体转动幅度较一级插入的大, 丙烯以二级插入方式较一级插入过程的能垒更高. 同时, 分析催化剂茂环上甲基H原子与烯烃甲基上H原子的距离可以发现, A-TS(2,1)si的H—H距离小于A-TS(2,1)re的H—H距离, 说明A-TS(2,1)si的烯烃甲基处于更加拥挤的环境中, 因此(2,1)si插入的能垒更高. 比较2个一级插入过渡态的结构信息[A-TS(1,2)si: C1—H5=0.111 nm, Ti—H5=0.229 nm, ∠Ti—C1—H5 =78.5°; A-TS(1,2)re: C1—H5=0.110 nm, Ti—H5=0.237 nm, ∠Ti—C1—H5 =80.7°]可知, A-TS(1,2)si的Ti—H5键更短, ∠Ti—C1—H5更小, 因此其抓氢(α-agostic)效应更强. 这种强抓氢效应稳定了A-TS(1,2)si, 降低了反应能垒. 类似的, 也发现A-TS(2,1)si的抓氢效应强于A-TS(2,1)re. 因此, 综合以上因素, 在催化剂A催化的丙烯聚合链引发过程中, 催化剂的刚性桥联结构使单体二级插入的自由能垒更高, 丙烯更倾向与以甲基远离催化剂桥基端的一级插入模式完成配位插入过程, 其中最优势路径为(1,2)si路径.

Fig.3 Key parameters of geometrical structures and free energies information of transition states(TSs) of four olefin insertion modes in the chain initiation process of propylene polymerization catalyzed by catalyst A Bond lengths are in nm, bond angles and dihedral angles in degree, energies are in kJ/mol, values in parentheses are zero-point energies.
如表1所示, 催化剂B和C催化丙烯聚合反应中烯烃插入过程的优势路径也是一级插入. 由于与萘环配体的强烈排斥作用, 丙烯无法以(2,1)re的方式同催化剂C完成配位插入过程. 此外, 催化剂B和C的二级插入活化位垒分别为84.2和98.4 kJ/mol, 比一级插入分别高了15.1和24.7 kJ/mol(一级插入的自由能位垒分别为69.1和73.7 kJ/mol). 3种催化剂催化烯烃聚合反应的链引发步骤的最优势路径都是(1,2)插入模式, 表现出一致的区位选择性, 这与它们具有共同的桥联空间结构有关.

Table 1 Free energy information of the chain initiation process of propylene polymerization catalyzed by catalyst A, B and C
* NA means not available.
2.2立体规整性
由上述对桥联茂钛配合物催化丙烯聚合反应链引发步骤的研究可见, 3种催化剂都表现出了较强的反应区位选择性. 由于(1,2)si和(1,2)re插入路径的反应能垒相差不大, 催化剂催化丙烯聚合反应开始并没有表现出明显的立体选择性, 因此对链引发步骤后续的第2、 第3个丙烯分子单体的配位插入过程的研究就尤为必要. 以催化剂B为例, 链引发过程完成后, 烷基配合物3(1,2)re的碳链绕Ti-Cα轴进行了旋转, 碳链摆动到另一侧得到烷基配合物4(1,2)re, 配位点也发生了迁移. 第2、 第3丙烯分子单体的配位插入过程的能量示意图见图4, 能量基点为4(1,2)re和丙烯的能量之和. 图中4→5→TS56→6为第2分子单体配位插入过程, (1,2)si和(1,2)re途径的反应自由能位垒分别为85.4和79.1 kJ/mol, 后者[(1,2)re途径]更具优势. 随着碳增长链的摆动, 烯烃配位点发生迁移, 7→8→TS89→9为第3丙烯分子单体的配位插入过程, (1,2)si和(1,2)re途径的自由能位垒分别为74.9和90.0 kJ/mol, (1,2)si路径为优势的反应途径. 比较第2、 第3丙烯分子单体的链传递过程, 增长链的异丁基由于催化剂配体环境的立体效应远离芴基配体(见图4中的4). 由于较小的立体效应, 丙烯单体采取甲基远离异丁基的空间取向进行配位插入的反应路径更占优势, 即第2个丙烯分子单体的(1,2)re途径; 同理, 在第3个丙烯分子单体的配位插入反应步骤中, 丙烯采取甲基远离生成的2,4-二甲基戊基的空间取向进行配位插入的反应路径更具优势, 即(1,2)si方式. 2个相邻插入单体在链传递步骤中具有相反的插入立体关系(re→si), 因此, 催化剂B过渡金属中心、 配体和增长链共同构成的立体环境使两相邻的插入单体的立体构型始终相反, 最终得到间规立构的聚丙烯链段.

Fig.4 Free energy profiles of the 2nd and 3rd monomer insertion processes of propylene polymerization catalyzed by catalyst B The values in parentheses are zero-point energies.
催化剂A和C催化丙烯聚合链传递步骤的反应自由能如表2所示. 催化剂A的结构同催化剂B的结构类似, 其区别在于茂环配体的立体位阻效应比芴配体要小得多, 反应活性空间相对宽松, 在催化丙烯聚合反应的链传递过程中, 碳增长链的空间取向并没有受到限制. 虽然催化剂A催化丙烯聚合反应的链传递过程中的第2和第3分子单体插入优势路径均为re路径, 但由于催化剂A的增长链摆动效应比催化剂B要容易, 因此实验中其催化产物主要是无规立构的[31]. 催化剂C催化丙烯聚合的链传递过程中, 碳增长链总是指向远离茚配体的苯环一侧, 因此, 配位丙烯的甲基会远离增长链以减少与配体的立体排斥作用. 由于催化剂C具有C2对称性, 单体插入的优势路径均为re路径, 相邻的2个单体配位插入的立体选择性相同, 因而得到等规立构的链段. 同时, 需要注意的是, 由于催化剂C的茚配体具有更强的立体效应, 2条路径的能垒差相对催化剂A和B更大, 说明催化剂C的立体定向能力更强.

Table 2 Free energy information of the chain propagation process of propylene polymerization catalyzed by catalysts A, B and C
3 结 论
对3种硅桥联的钛茂催化剂催化丙烯聚合的链引发和链传递过程进行了密度泛函理论研究. 计算结果表明, 催化剂A, B和C的链引发优势路径分别为(1,2)re和(1,2)si的一级插入反应模式, 其插入的区位选择性与它们共同具有的刚性硅桥联结构密切相关. 在链引发步骤后, 烯烃插入的链增长过程中, 由于催化剂结构对称性的不同, 催化剂过渡金属中心的配体、 增长链构成的立体构象对生成聚烯烃产物的立体规整性具有非常重要的影响. 具有Cs对称性的A和B催化丙烯聚合分别得到无规立构和间规立构的聚烯烃产物, 其中A的增长链的摆动效应是其无规立构的主要原因. 而具有C2对称性的C催化丙烯聚合得到等规立构聚烯烃产物与实验结果一致. 本文从理论角度研究了硅桥联的钛茂催化剂催化α-烯烃聚合反应过程中的区位选择性和立体选择性及其影响因素, 为设计改性潜在高选择性的聚烯烃催化剂提供了一定的理论基础.
支持信息见http://www.cjcu.jlu.edu.cn/CN/10.7503/cjcu20160271.
[ 1 ]Zhao Y., Hu Q., Li D. X.,ChinaSyntheticResinandPlastics, 2003, 20(5), 67—71(赵燕, 胡清, 李德旭. 合成树脂及塑料, 2003, 20(5), 67—71)
[ 2 ]Guo L. H., Chen C. L.,Sci.China:Chem., 2015, 58(11), 1663—1673
[ 3 ]Liu H., Liu L., Wang F., Jia X. Y., Bai C. X., Zhang C. Y., Zhang X. Q.,Chem.J.ChineseUniversities, 2014, 35(6), 1336—1342(刘恒, 刘李, 王凤, 贾翔宇, 白晨曦, 张春雨, 张学全. 高等学校化学学报, 2014, 35(6), 1336—1342)
[ 4 ]Yang X., Stern C. L., Marks T. J.,J.Am.Chem.Soc., 1994, 116(22), 10015—10031
[ 5 ]Woo T., Fan L., Ziegler T.,Organometallics, 1994, 13(6), 2252—2261
[ 6 ]Woo T. K., Margl P. M., Ziegler T., Blöchl P. E.,Organometallics, 1997, 16(15), 3454—3468
[ 7 ]Yang F., Lv Z. B., Zhao Y. Q.,Contemp.Chem.Ind., 2014, 43(6), 973—974(杨帆, 吕振波, 赵瑛祁. 当代化工, 2014, 43(6), 973—974)
[ 8 ]Xu X. X., Yi J. J., Jing Z. H.,ActaPolymericaSinica, 2001, 1(5), 683—686(许学翔, 义建军, 景振华. 高分子学报, 2001, 1(5), 683—686)
[ 9 ]Qian C. T., Wang C. H., Chen Y. F.,ActaChimicaSinica, 2014, 72(8), 883—905(钱长涛, 王春红, 陈耀峰. 化学学报, 2014, 72(8), 883—905)
[10]Resconi L., Cavallo L., Fait A., Piemontesi F.,Chem.Rev., 2000, 100(4), 1253—1346
[11]Kaminsky W.,Catal.Today, 2000, 62(1), 23—34
[12]Schellenberg J., Tomotsu N.,Prog.Polym.Sci., 2002, 27(9), 1925—1982
[13]Jordan R. F.,Adv.Organomet.Chem., 1991, 32, 325—387
[14]Pellecchia C., Proto A., Longo P., Zambelli A.,Makromo.Chem.,RapidCommun., 1991, 12(12), 663—667
[15]Lohrenz J. C., Woo T. K., Fan L., Ziegler T.,J.Organomet.Chem., 1995, 497(1), 91—104
[16]Corradini P., Cavallo L., Guerra G.,MolecularModelingStudiesonStereospecificityandRegiospecificityofPropenePolymerizationbyMetallocenes, John Wiley & Sons Ltd., Chichester, 2000
[17]Correa A., Galdi N., Izzo L., Cavallo L., Oliva L.,Organometallics, 2008, 27(6), 1028—1029
[18]Kawamura-Kuribayashi H., Koga N., Morokuma K.,J.Am.Chem.Soc., 1992, 114(22), 8687—8694
[19]Cruz V. L., Ramos J., Martinez S., Muoz-Escalona A., Martinez-Salazar J.,Organometallics, 2005, 24(21), 5095—5102
[20]Chai J. D., Head-Gordon M.,J.Chem.Phys., 2008, 128(8), 084106
[21]Frisch M. J., Trucks G. W., Schlegel H. B., Scuseria G. E., Robb M. A., Cheeseman J. R., Scalmani G., Barone V., Mennucci B., Petersson G. A., Nakatsuji H., Caricato M., Li X., Hratchian H. P., Izmaylov A. F., Bloino J., Zheng G., Sonnenberg J. L., Hada M., Ehara M., Toyota K., Fukuda R., Hasegawa J., Ishida M., Nakajima T., Honda Y., Kitao O., Nakai H., Vreven T., Montgomery J. A. Jr., Peralta J. E., Ogliaro F., Bearpark M., Heyd J. J., Brothers E., Kudin K. N., Staroverov V. N., Keith T., Kobayashi R., Normand J., Raghavachari K., Rendell A., Burant J. C., Iyengar S. S., Tomasi J., Cossi M., Rega N., Millam J. M., Klene M., Knox J. E., Cross J. B., Bakken V., Adamo C., Jaramillo J., Gomperts R., Stratmann R. E., Yazyev O., Austin A. J., Cammi R., Pomelli C., Ochterski J. W., Martin R. L., Morokuma K., Zakrzewski V. G., Voth G. A., Salvador P., Dannenberg J. J., Dapprich S., Daniels A. D., Farkas O., Foresman J. B., Ortiz J. V., Cioslowski J., Fox D. J.,Gaussian09,RevisionD.01, Gaussian Inc., Wallingford CT, 2010
[22]Wang M., Zhang X., Chen Z., Tang Y., Lei M.,ScienceChinaChemistry, 2014, 57(9), 1264—1275
[23]Lei M., Wang Z., Du X., Zhang X., Tang Y.,J.Phys.Chem.A, 2014, 118(39), 8960—8970
[24]Ma X., Tang Y., Lei M.,DaltonTrans., 2014, 43(30), 11658—11666
[25]Lei M., Pan Y., Ma X.,Eur.J.Inorg.Chem., 2015, 2015(5), 794—803
[26]Ma X., Lei M., Liu S.,Organometallics, 2015, 34(7), 1255—1263
[27]Li H., Ma X., Lei M.,DaltonTrans., 2016, 45, 8506—8612
[28]Li L., Pan Y., Lei M.,Catal.Sci.Technol., 2016, 6, 4450—4457
[29]Zhang Y. W., Ma X. L., Zhang X., Lei M.,ActaChim.Sinica, 2016, 74(4), 340—350(张益伟, 马雪璐, 张欣, 雷鸣. 化学学报, 2016, 74(4), 340—350)
[30]Dahlmann M., Erker G., Nissinen M., Fröhlich R.,J.Am.Chem.Soc., 1999, 121(12), 2820—2828
[31]Resconi L., Camurati I., Grandini C., Rinaldi M., Mascellani N., Traverso O.,J.Organomet.Chem., 2002, 664(1), 5—26
(Ed.: Y, Z, S)
† Supported by the Major Science Foundation of Beijing Institute of Fashion Technology, China(No.2014A-01).
Theoretical Studies on the Regioselectivity and Stereoselectivity of the Olefin Polymerization Catalyzed by Bridged-metallocene Complexes†
TANG Yanhui1*, WANG Zhidong2
(1.SchoolofMaterialsScienceandEngineering,BeijingInstituteofFashionTechnology,Beijing100029,China; 2.StateKeyLaboratoryofChemicalResourceEngineering,CollegeofScience,BeijingUniversityofChemicalTechnology,Beijing100029,China)
The reaction selectivity of olefin polymerization catalyzed by three different bridged-metallocene complexes[Me2SiN(Me4Cp)TiCl2, Me2SiCpFluTiCl2and Me2SiInd2TiCl2] were studied with density functional theory. The calculation results show that the ligand of the metalloence complex creates special stereo environment which leads to unique catalytic properties. There are two ways for olefin insertion, primary and secondary insertion, in the chain initiation and chain propagation steps of propylene polymerization catalyzed by bridged-metallocene catalyst. The calculation results suggest that bridged-metallocene catalysts show a preference of primary insertion way in chain initiation step of propylene polymerization, and we believe this regioselectivity is resulted from the rigid structure of catalyst. In the subsequent chain growth step, the catalysts start to show their stereoregularity, and the product of polymerization isatac-,syn-,iso-PP, respectively. By studying the interactions of the monomer, the ligand and the growing chain, we revealed the origin of the regioselectivity and stereospecificity of bridged-metallocene catalyst and provide theoretical support for design of tailor made metallocene catalyst.
Bridged-metallocene complex; Olefin polymerization; Regioselectivity and stereoregularity; Reaction mechanism; Theoretical study
10.7503/cjcu20160271
2016-04-22. 网络出版日期: 2016-09-23.
北京服装学院重点科研项目(批准号: 2014A-01)资助.
O643.31
A
联系人简介: 唐艳辉, 女, 讲师, 主要从事计算催化化学研究. E-mail: clyyanhuitang@bift.edu.cn
猜你喜欢
大众文艺(2022年16期)2022-09-07
中国民族美术(2021年4期)2021-07-14
四川轻化工大学学报(自然科学版)(2021年1期)2021-06-09
中国特种设备安全(2019年9期)2019-12-03
农药科学与管理(2019年5期)2019-08-13
当代陕西(2019年6期)2019-04-17
画刊(2018年2期)2018-03-06
化工管理(2016年31期)2016-12-15
中国工程咨询(2015年2期)2015-02-14
外语学刊(2014年3期)2014-12-03
