
长QT综合征的诊断及治疗
2016-10-28 06:50胡宇才阮燕菲
中西医结合心血管病杂志(电子版) 2016年17期
胡宇才,阮燕菲*
(首都医科大学附属北京安贞医院,北京 100029)
长QT综合征的诊断及治疗
胡宇才,阮燕菲*
(首都医科大学附属北京安贞医院,北京 100029)
1 定义
长QT间期综合征(LQTS)是以心电图上QT间期延长为基础而诊断的,可伴T波及ST段改变,临床上则表现为易发生恶性室性心律失常如尖端扭转型室速(Torsades de Pointes,Tdp)、室颤等,患者可有反复发作的黑矇、晕厥,甚至猝死。
2 LQTS的病理生理基础及病因学分类
2.1 病理生理基础
各种LQTS共同的病理生理基础是由于先天性或获得性原因使心肌细胞膜上的离子通道功能异常,如编码钠和钾离子通道亚单位的基因发生突变导致心肌复极时内向钠通道失活延迟或钾离子外流延缓,导致动作电位复极时间延长和复极离散度增大,对应的心电图变化即为QT间期延长和在此基础上发生的室性心律失常。
2.2 病因学分类及其表现
2.2.1 遗传性LQTS 即具有遗传背景的LQTS,是由于染色体异常或基因突变导致心肌细胞膜上离子通道功能障碍所致,常呈家族性发病。遗传性LQTS在临床上又分为两型:Jervell-Lange-Nielsen(JLN)综合征和Romano-Ward(RW)综合征。
2.2.1.1 JLN综合征
Jervell-Lange-Nielsen(JLN)综合征为少见类型,是一种伴有神经性耳聋的先天性长QT 间期综合征,属于常染色体隐性遗传病,1957 年首先由Jervell 和Lange、Nielsen 报告,故称为JLN综合征,又称为“聋心综合征”。临床上以先天聋哑、QT 间期延长、多形性室性心动过速、尖端扭转性室性心动过速以及发作性晕厥、心脏性猝死为特征。尽管JLN综合征为一种罕见疾病,患病率为1.6~6/100万,但耳聋儿童中本病发病率为0.25~1%。JLN综合征由编码心肌细胞钾通道的KCNQ1或KCNE1基因突变所致,属常染色体隐性遗传,父母双方各带一个相同或不同的突变并同时把突变传给子代才会患病,所以病例较少。患儿心电图上QT间期常大于500ms,伴有双侧严重的先天性感觉性听力丧失。该型患者90%发生过心脏事件,50%在三岁即出现症状(晕厥),比其它亚型LQT综合征患者发病年龄要早,病人首次出现晕厥后10年内病死率接近50%。仅少数JLN综合征患者对常规β受体阻滞剂和左侧心交感神经节切除术(LCSD)治疗有效;未经治疗的JLN综合征患者一半以上在15岁前死亡。
2.2.1.2 RW综合征
Romano-Ward(RW)综合征占遗传性LQTS的大部分,是由编码心肌细胞钾通道或钠通道的KCNQ1、KCNH2、KCNE1、KCNE2或SCN5A基因突变所致的一种常染色体显性遗传病,心电图表现为QT间期延长、T波异常以及频繁的发生尖端扭转室速(TdP),TdP有自限性,但少数情况下也会蜕变为室颤导致患者猝死。患者听力正常,可反复发作晕厥,晕厥多在运动或情绪激动时发作,少数在睡眠或休息时发生,发作前常无先兆。携带有与RW综合征相关的突变基因患者中大约50%在一生中会至少有一次晕厥发作,首次发病可以在从婴儿时期至中年间的任何时候,取决于家族的基因类型,最常见是从十几岁至二十岁。根据突变基因的不同,RW综合征可施予β受体阻滞剂或钠通道阻滞剂治疗,左侧心交感神经节切除术对部分患者也有效。
2.2.2 获得性LQTS
即继发于某些后天性、外源性因素的LQTS,如(1)药物—包括抗心律失常药(奎尼丁,索他洛尔,胺碘酮,依布利特)、三环类抗精神病药、抗微生物药物(红霉素,氯奎,金刚胺)、抗组胺药(阿司咪唑)以及西沙比利、砷剂、有机磷等;(2)电解质紊乱--低钾,低钙,低镁;(3)严重缓慢性心律失常;(4)脑血管疾病--脑卒中、脑外伤等;(5)心脏疾病--心肌炎、心肌缺血等;(6)甲状腺功能减退、低温、自主神经性疾病等。获得性LQTS多为可逆性,去除诱因后QT间期可恢复正常。
随着近年来对LQTS基因型与表型关系研究的深入,发现遗传性和获得性LQTS的病例有部分重叠。一些散发的LQTS病例,经基因诊断存在LQTS的遗传背景但常规心电图上QT间期并不延长,这些病例仅仅只在有上述获得性LQTS诱因存在时才表现出QT间期延长及相关的心律失常。
3 遗传性LQTS的诊断标准
除了排除获得性LQTS以外,影响QT间期的因素还有很多,如心率、T波终点的确定等,而且所谓“QT间期正常值”也是基于统计学数据的一个区间值(表1),在诊断遗传性LQTS时还必须个体化。PeterJ.Schwartz于1993年提出的计分法(表2)用于临床上诊断遗传性LQTS,敏感性和特异性均较高。按照该计分法,≤1分者,LQTS的诊断可能性小;2~3分者,诊断可疑;≥4分者,诊断肯定。
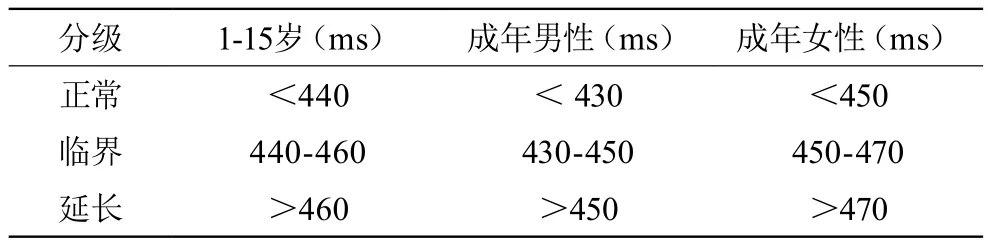
表1 不同年龄、性别的QT间期正常与异常值范围

表2 Schwartz评分法长QT综合征诊断标准
4 遗传性LQTS分型
随着分子遗传学研究的进展,很多(75%)遗传性LQTS的基因突变类型及其相关离子通道功能改变已被阐明,即基因型-表型关系已经明确,并依此对遗传性LQTS进行了分型(按阿拉伯数字排序),目前已知的有13型-LQTS1、LQTS2、LQTS3…LQTS13(表3)。其中LQTS1、LQTS2和LQTS3占所有已确定基因型遗传性LQTS病例的95%。
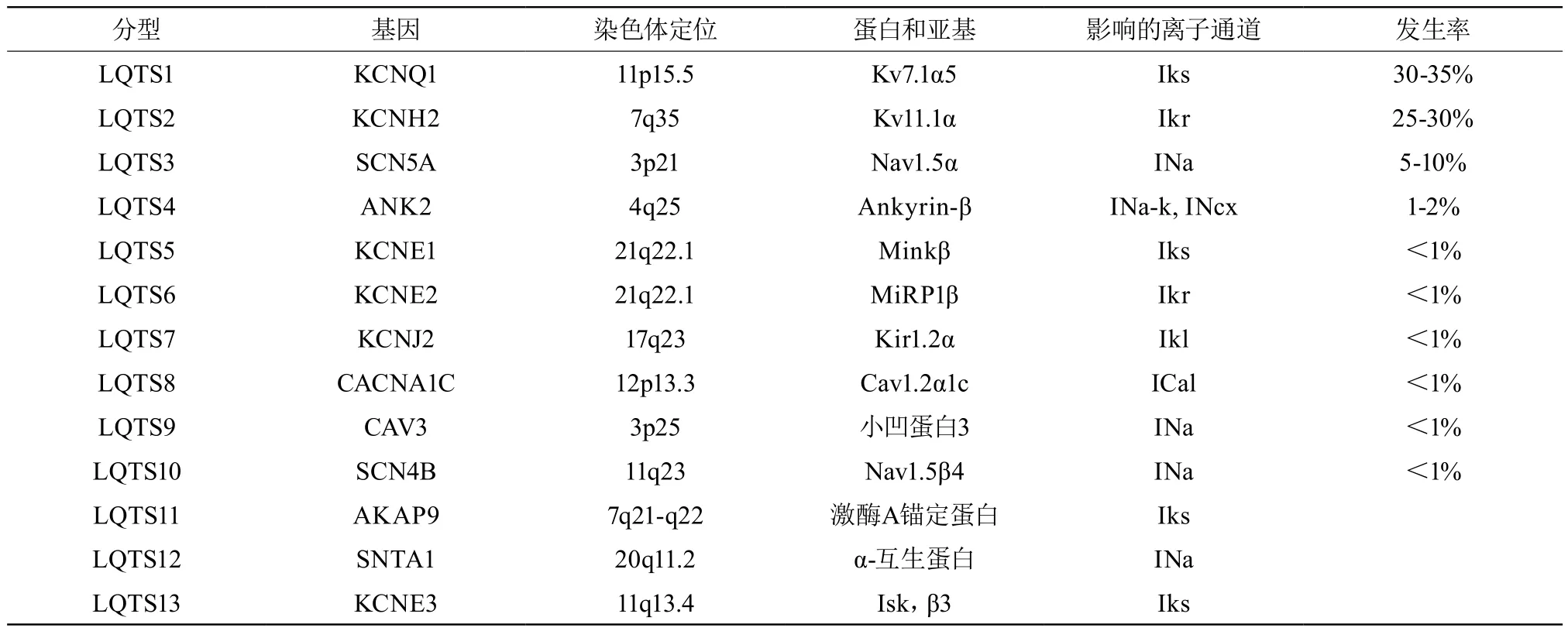
表3 遗传性长QT综合征的分型
4.1 LQTS1类型特点
LQTS1患者典型的心电图表现为基底宽大的单向T波,QT间期中度延长,QTc≥500 ms者发生心脏事件的危险性更高。LQTS1患者的心律失常事件大多数由活动诱发,游泳是主要诱因之一。LQTS1对β受体阻滞剂治疗反应很好。
LQT1的心电图特征:T波基底部增宽,有4种形态:a.“婴儿型”T波:T波为非对称性高耸、基底增宽;b.宽基底T波:T波基底增宽,起始点不明显;c.T波正常出现,形态正常;d.T波延迟出现,形态正常(图1)。
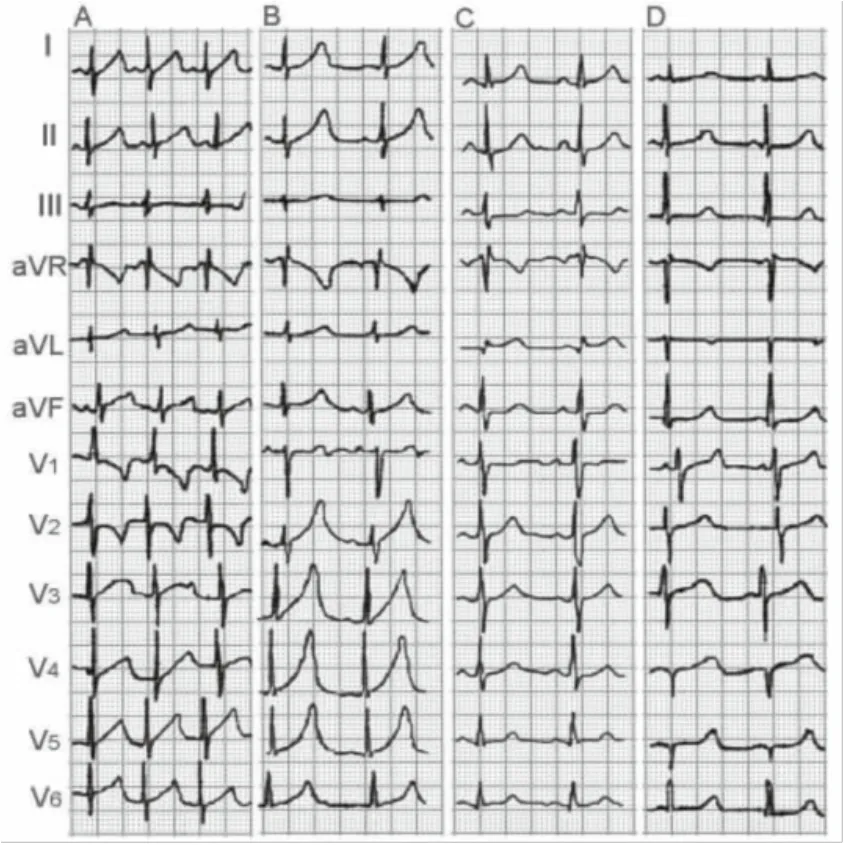
图1 遗传性长综合征1型(LQTS1)的四种心电图表现
LQTS1是遗传性LQTS的最常见类型,在确定基因型的病例中占50%。缓慢激活延迟整流钾通道(IKs)的功能缺失是LQTS1的病理生理基础。1996年,中国学者Wang Q.等首先克隆了位于染色体11p15.5的KCNQ1基因。它由17个外显子组成,长约400kb,编码IKs的α亚基。KCNQ1通道是由4个α亚基相互作用形成的四聚体。α亚基含有6个跨膜片段(S1~S6)和一个埋藏在脂质双层膜中的孔。S4含有许多带正电荷的氨基酸,是通道的电压感受器,S5~S6之间的连接区域形成离子的传导通路。氨基(n-)端和羧基(c-)端位于细胞内。KCNQ1突变有错义突变、无义突变、剪接突变、氨基酸缺失、移码突变等,大部分突变发生在跨膜区或细胞内部分。基因突变引起IKs的α亚单位功能受损,IKs减少,动作电位时程延长,心电图上表现为QT延长。
4.2 LQTS2类型特点
这一类型患者的大多数心律失常事件由情绪激动诱发,铃声刺激是LQTS2患者的特异性触发因素。LQTS2患者对β受体阻滞剂的反应不及LQTS1患者,因此对QT间期显著延长或反复晕厥的高危患者,可考虑预防性植入ICD并联合使用β受体阻滞剂。
LQT2的心电图特征:T波振幅低而有切迹(或双峰),有6种形态:a.明显T波双峰;b.微小的T波双峰,第二峰出现于T波顶部;c.微小的T波双峰,第二峰出现于T波降支;d.振幅低平的双峰T波,第二峰略高;e.振幅较高的双峰T波,第二峰更高;f.略高于基线的双峰。(图2)
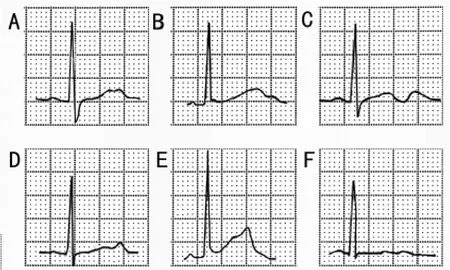
图2 遗传性长综合征2型(LQTS2)的六种心电图表现36
LQTS2的致病基因为HERG( human“ether-a-gogo”related gene),位于7q36.1,由4个α亚单位组成,每个亚单位有6个跨膜片段,1个孔区及氨基和羧基末端,n和c末端均位于细胞内,跨膜片段s5、s6及连接两者的胞内肽段共同构成通道孔的结构域,跨膜区与孔区是突变的好发位点。HERG编码快速激活的延迟整流钾通道(IKr)的α亚基,IKr是心室肌细胞动作电位复极期中的主要外向电流。HERG基因突变则导致IKr通道失活,外向钾电流减少或消失,引起心肌复极时间(QT间期)延长。HERG突变涉及通道的各个区域,目前将HERG突变致LQTS2的机制归纳为:①合成异常;②细胞内运输异常;③通道门控异常;④通道离子选择性异常。一种突变可通过4种机制中的一种或数种发挥作用,其中合成异常和细胞内运输异常是HERG基因错义突变导致LQTS2的最常见机制。
4.3 LQTS3类型特点
LQTS3的患病数在所有确定基因型的LQTS患者中约占10-15%。LQTS3的典型心电图表现为ST段延长和小而尖的T波,大多数心律失常事件发生于休息状态或睡眠中。患者对β受体阻滞剂反应差,部分患者使用钠通道阻滞剂可减少晕厥或TdP发作,高危的LQTS3患者应植入ICD。
LQT3的心电图特征:ST段延长,T波延迟出现,婴幼儿期易发生2:1房室阻滞,T波形态有2种:a. T波延迟出现,高耸或呈双相;b. T波非对称性高耸。须注意的是在同一家系中,长QT综合征患者之间,T波形态可有重叠;而不同致病基因之间T波形态也有重叠。(图3)
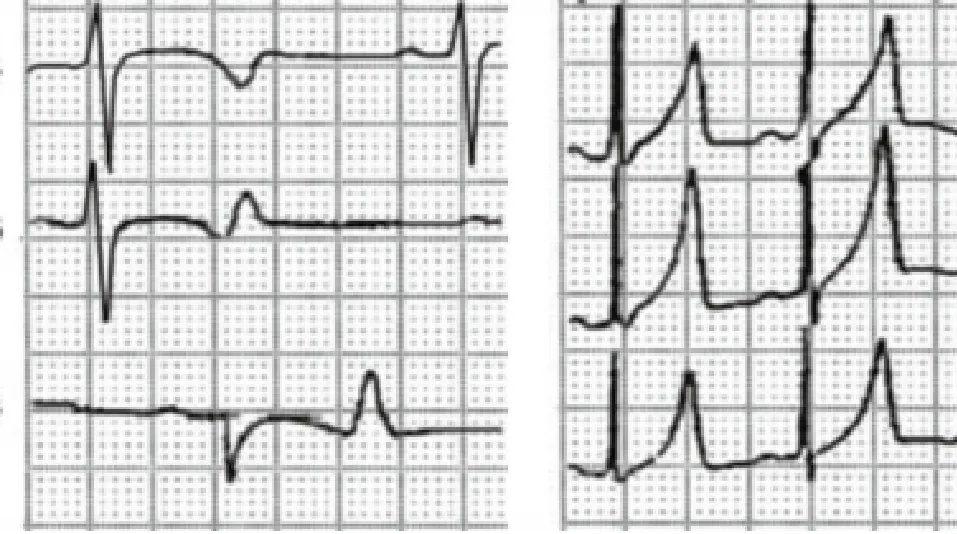
图3 遗传性长综合征3型(LQTS3)的心电图特征
LQTS3的致病基因是编码心脏门控钠通道的SCN5A,定位于3p21,由28个外显子组成,编码一由2016个氨基酸组成的蛋白质。该蛋白在细胞膜上形成4个结构类似的同源结构域,每个区域由6个跨膜片段组成,形成钠通道α单位;其中s5和s6片段之间的连接环构成通道孔,通道孔具有不对称结构,选择性通过Na离子。s4片段为通道的电压感受器,当细胞膜电位除极时可使s4片段发生跨膜移动,激活钠通道产生钠电流。SCN5A基因突变可改变钠通道的正常结构,进而改变钠通道的功能,并导致心律失常的发生。突变使钠通道失活延迟,2相的Na电流持续不失活,使复极和动作电位的时程延长。由于动作电位时程的异常延长,使早期后除极及触发活动增加,并易诱发尖端扭转型室速。研究表明钠通道的失活延迟与心率减慢有关,因而LQTS3患者多在心率缓慢或睡眠时发生心律失常事件。
5 LQTS心脏以外的表现
目前发现LQTS7与LQTS8患者有心脏以外的临床表现,有助于鉴别诊断。
5.1 Andersen-Tawil综合征
即LQTS7,是一种常染色体显性遗传病,其特征为周期性麻痹、QT间期延长伴心律失常、机体畸形三联征,其中典型的畸形包括宽眼距、小下颌、低耳廓、第5指(趾)弯曲、第2和3指(趾)并指(趾)、身材矮小以及脊柱侧凸等。70%的LQTS7与KCNJ2基因突变有关,后者使内向整流钾电流(KIr2.1)减小,引起延迟后除极和心律失常,心电图上可表现为QT间期轻度延长、宽大U波等。Andersen-Tawil综合征患者在幼年或少年时期出现心脏症状(心悸、晕厥)和/或运动障碍。
5.2 Timothy综合征
即LQTS8,是编码电压门控的钙离子通道基因(CACNA1C)的突变所致的一种较罕见的LQTS临床表型。由于离子通道失活钝化,导致钙超载从而引起外周组织发育和功能异常,同时伴有心电图改变和心律失常。心电图上可见显著延长的QT间期、房室传导阻滞和巨大的T波。心外表现中最突出的是并趾畸形,一部分Timothy综合征儿童还可有免疫缺陷、认知障碍、间隙性低血糖以及孤独症。Timothy综合征恶性程度较高,大部分患儿死亡时的平均年龄为2.5岁。
6 LQTS的治疗原则
获得性LQTS通常不需要长期治疗,去除引起QT间期延长的原因后,QT间期多可恢复正常。
遗传性LQTS患者必须进行长期预防性的治疗以降低猝死的风险。β受体阻滞剂能显著减少心律失常事件的发生,在LQTS1患者疗效更为明显,对于有明显心动过缓的患者,可在安装了永久起搏器后维持β受体阻滞剂治疗。不能耐受β受体阻滞剂或治疗效果不佳的患者,左侧心交感神经节切除术(LCSD)可作为二线治疗。对于接受足量β受体阻滞剂和LCSD治疗或安装了永久起搏器后仍反复发生晕厥/Tdp的患者应考虑植入ICD,ICD虽不能预防恶性心律失常的发生,但可有效预防猝死。
由于遗传性LQTS的治疗和预后有着鲜明的基因特异性,基因学结果指导下的个体化治疗策略将在很大程度上提高LQTS患者及其家族成员的治疗效果。尽管目前进行基因测定和分型的价格还很昂贵,但随着分子生物学的发展和商业运作的参与,遗传性LQTS的基因诊断和治疗必定成为一种趋势而得到广泛推广应用。
读者・作者・编者
本刊投稿注意事项
《中西医结合心血管病杂志》杂志来稿内容应遵循本刊稿约要求和各栏目说明,除内容要求以外,特提醒作者注意以下情况。
1.需要在文中表达“作者简介”或“通讯作者简介”的,请提供作者的相关信息,一般格式为“姓名(出生年月-),性别,籍贯,最高学位,职称,研究方向或者工作方向等”;若在科研工作中取得了突出成绩,可适当详细介绍,一般不超过100字。对于缺乏实质内容介绍的,如仅表达了工作单位的,将不予在刊出时标明。
2.署名及排序由作者在投稿时确定,投稿后一般不得改动。通讯作者非第一作者时,须注明通讯作者;不注明者,默认第一作者为通讯作者。有多个作者而非同一单位的,单位表述必须清楚、准确,中英文表述均应使用官方名称。
3.基金来源的稿件需注意:①有效性。基金来源的稿件在正文中的标注应与其基金证明文件一致(要求随稿提供传真件、复印件等证明材料),确保准确;禁止明显与该论文内容无关的虚假标注。②完整性。在文中的“基金项目”标注处,须完整标示其项目来源、课题名称及编号。完整表述如:广东省中医药局科研课题:蓝菊搞病毒口服液工艺及质量标准研究(项目号:2009121)。
4.本刊为黑白印刷,文中若有彩色图片的,请作者自行处理,以黑白印刷不影响图片表达为度。

R331.3
A
ISSN.2095-6681.2016.17.005.04
阮燕菲,E-mail:ruanyanfei@hotmail.com
胡宇才,男,35岁,医学博士在读,现为河南中医药大学第一附属医院心脏中心主治医师,兼任中国生物医学工程学会起搏与电生理分会会员,河南省医师学会影像技术专业委员会委员,河南省中医药学会心血管疾病专业委员会委员,河南省中医药学会络病专业委员会委员,河南省中西医结合学会循证医学专业委员会委员。研究方向为中西医结合治疗心血管疾病的基础和临床研究,擅长心、脑血管系统疾病的影像诊断及心律失常电生理检查与射频消融治疗技术。
猜你喜欢
中国现代医生(2022年19期)2022-11-04
中国肿瘤临床(2022年14期)2022-08-09
昆明医科大学学报(2022年4期)2022-05-23
中国典型病例大全(2022年7期)2022-04-22
中国听力语言康复科学杂志(2021年6期)2021-12-21
现代仪器与医疗(2021年2期)2021-07-21
心电与循环(2021年3期)2021-06-03
世界科学技术-中医药现代化(2021年12期)2021-04-19
心电与循环(2021年1期)2021-02-05
中国循证心血管医学杂志(2018年8期)2018-01-16
