
超价分子中的d轨道
2016-10-13 01:22:05陈天阳范如本陈翔宇崔智昊北京大学化学与分子工程学院北京100871
大学化学 2016年2期
陈天阳 范如本 陈翔宇 崔智昊(北京大学化学与分子工程学院,北京100871)
·未来化学家·
超价分子中的d轨道
陈天阳范如本陈翔宇崔智昊*
(北京大学化学与分子工程学院,北京100871)
通过对一些典型超价分子进行计算和分析,得出了超价分子“d轨道参与”(即外层d轨道杂化和d-p π键概念)不尽合理的结论,并提出了能与实验事实相符的解释方法。此外,本文还阐述了计算化学中基组d函数与d轨道的关系:二者并不等价。
超价分子;d轨道;杂化轨道理论;d-p π键;极化基组
www.dxhx.pku.edu.cn
超价分子(hypervalent molecule)一词产生于经典的Lewis八隅体结构理论,用来描述那些主族元素中心原子形式上拥有超过8个电子的化合物。超价分子的一个确切定义由Musher[1]给出:由15-18族的高价态原子作为中心原子形成的分子。这样的例子很常见,如PCl5、SF6、XeF2等分子以及SO2-4等离子。
为了解释这些超价分子的成键,杂化轨道理论给出了外轨型杂化的解释,比如认为SF6是sp3d2杂化,核心是引入了外层的d轨道参与杂化。之后为了解释POF3等分子的结构,引入了d-p π键来解释键长的缩短。但是这些外层的d轨道究竟是否参与了成键,对分子的几何构型是否有影响呢?本文以d轨道为主线,对几个典型的超价分子进行了计算和讨论,并提出了一些新的理论解释。
1 外层d轨道杂化的不合理性
1.1一个经典分子:SF6
SF6是一个经典的超价分子。杂化轨道理论认为:S的1个3s轨道、3个3p轨道、2个3d轨道形成sp3d2杂化轨道与F的p轨道叠加成键,并根据共价键的饱和性解释了SF6的水解稳定性。形成上述杂化轨道的理论基础是:S是第三周期元素,有d轨道;利用上述几个轨道杂化,可以满足正八面体的对称性。
尽管杂化轨道理论看似较好地解释了SF6的几何构型,但是从分子轨道的角度来看,这种说法是不准确的。分子轨道理论认为,如果2个原子轨道能形成有效的成键轨道,必须满足下列条件:(1)轨道对称性匹配;(2)轨道能级相近;(3)轨道之间能够有效重叠。其中条件(2)是这里讨论的重点。一般认为,能级差距大于10 eV的原子轨道不能有效地形成成键分子轨道,而是成为非键轨道,并几乎保持原来原子轨道的性质。
文献[2]显示,S的3d轨道轨道能的负值为1.94 eV,而F的2p轨道为17.4 eV,能级显然低于S的3d轨道10 eV以上(参看表1中的粗体数据),这说明在SF6中,3d轨道几乎没有参与成键。
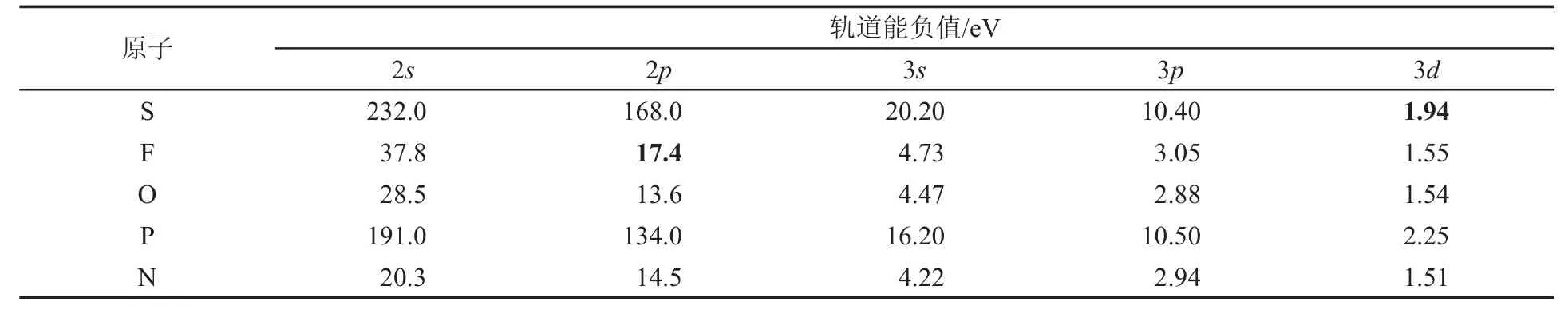
表1 本文中涉及到的一些元素的轨道能负值[2]
不仅从原子轨道能量上可以简单定性地分析出d轨道参与的有限性,而且计算化学的结果给出的d成分分析和杂化轨道计算值也大不相同。如果SF6的中心原子S严格遵守sp3d2杂化,则d轨道成分为1/3。利用XMVB程序LANL2MB基组[3]进行基于价键理论的计算,可以得到如下计算结果:

由此可以看出,计算与杂化轨道理论的假设有着较大的差异,经过计算得出2条d轨道在成键时只贡献了0.038,远小于理论的1/3的比例。因此可以认为,在用杂化轨道理论解释超价分子的成键情况时引入d轨道是失败的。
那么应该如何解释SF6的成键情况呢?
分子轨道理论可以较好地阐明这个问题。问题的关键是,如何使S原子中的1个s轨道与3个p轨道形成6条键。在分子轨道理论的描述中引入了具有a1g,t1u和eg对称性的群轨道,其中a1g,t1u轨道分别和S的3s、3p轨道对称性匹配,组合成分子轨道,而eg轨道保持非键特性。故6条S—F键是由4条成键轨道以及2条非键轨道组成,平均键级为2/3。
此外,简单的多中心键的模型也可以解释SF6的成键情况。SF6中的6条S—F键共有3个空间取向,在其中一个轴向上是典型的2个S—F单键;在另外两个轴向上,S的充满的p轨道以及2个F半满的p轨道共同形成了3c-4e键。通过共振,每个S—F键的平均键级为2/3。此理论在没有引入d轨道参与成键的前提下较好地解释了SF6的成键情况。(不足的是,在SF6中S—F键长(156.1 pm)应比SF2中键级为1的S—F键要长;但实际上,SF2中S—F键长为159.1 pm,反而略长一点。)
1.2一个不可能用d轨道参与解释的例子:超价碳
根据Musher之前的定义,超价分子的中心原子并不包含碳。这是因为长期以来,形如A,B一类的超价碳原子一直没有被发现(图1)。但是五价硅以及六价硅是非常常见的,这从侧面说明了超价碳原子确实不易得到。对于C这类被环的刚性束缚而得以稳定化的五配位碳原子体系,已有不少报道[4-7]。2008年,一篇文献以类似的想法,构建了六配位的碳原子体系D和E(图2),并用DFT理论对其参数进行了模拟和优化[8]。
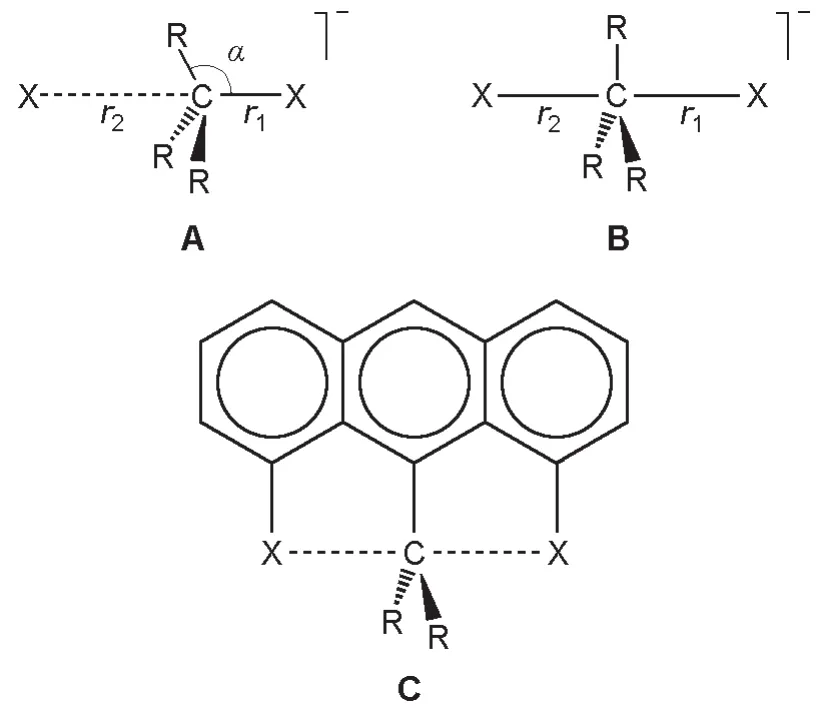
图1 几种类型超价碳的示意图
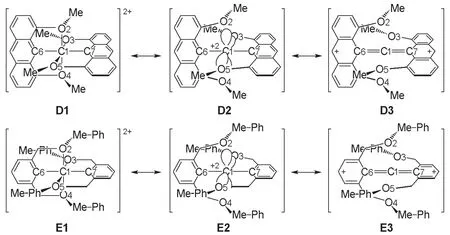
图2 超价碳D和E的结构
计算表明D并非超价分子,E才是超价体系。从共振式可以看出,D中正电荷通过菲环离域到了芳香体系内,且甲氧基有强烈的给电子效应,也帮助正电荷向芳环离域而不是向氧原子离域。因此,D中C1的行为更像丙二烯中的碳原子,键长和键能也支持这一点。而E中苯环离域能力不如菲环,且没有甲氧基,因此更利于体现超价特性。结合以上推理,我们提出以下假设:D中C6和C7的对位连接诸如硝基等强吸电子基,会使C1—C6和C1—C7间的距离变长,C—O距离变短,从而实现超价;反之,在E中同样位置连接给电子基团可以破坏超价体系。这些事实说明体系的形成是非常依赖中心原子所处的具体化学环境的,不能一概而论。
以上介绍了一类超价碳原子,这些体系超价的原因在于芳环系的刚性束缚,称为“分子脚手架”(molecular scaffold)。这还不是中心碳原子完全“自由”情况下的超价现象。因此,有的科学家根据双分子亲核取代反应的过渡态,设计并模拟了五配位碳原子[9],称为“冻结过渡态结构”(freezingSN2 TS)。
对于五配位三角双锥结构,Bickehaupt等提出了箱中球(ball-in-box)模型[10]和球中盘(disk-in-ball)模型[11]。第一个模型主要用于解释五配位硅化合物和五配位碳化合物稳定性的差异。第二个模型是在处理Ng-CL3-Ng(Ng表示稀有气体原子)体系时提出的。研究人员设计了合适的“盘子”(C(CN)3),可以适用这个模型(氢和卤素构成的盘子刚性不足,容易翻转)。取体积较大的碘原子作为径向原子(其他卤素原子与中心作用太强),优化结果表明这个体系是稳定的,具有理想模型下的实频吸收,C与I之间的共价键力常数为正值,而非推斥态,故C(CN)3I2-是稳定的五配位结构。
上述实例说明,碳原子在合适的条件下,完全可以像同族的硅原子那样形成五配位甚至六配位的结构,显然,对于碳原子,我们更不可能把原因归结为d轨道的参与。对电子结构的分析支持3c-4e键的说法,就如同SF6一样。至此,可以初步得出结论:d轨道不是超价的根源所在。对于碳等第二周期原子来说,难以超配位的原因还是由于几何尺寸不合适。
2 重新理解d-p π键
d-p π键是为了解释某些“单键”具有双键性质时引入的。其核心是p轨道和d轨道对称性匹配,可以具有一定程度的成键作用,从而使某键具有多重键的性质,例如对于POF3中P—O键长缩短的解释。但是,如前所述,3p轨道和3d轨道不满足“能级相近”原则,无法有效成键,更不要说能级更低的2p轨道了(表1),故需要对d-p π键进行重新理解。
2.1一个模型化合物:NOF3
NOF3是确实存在的一种化合物[12]。它像它的等电子体NF4+一样,似乎可以用Lewis结构式描述。但是“奇怪”的事情是:NOF3中N—O键长为115.8pm,有明显的多重键合。我们用Gaussian[13]DFT方法计算了NOF3和几个典型分子的N—O键长,将结果列于表2。根据表2可以推断N—O键级约为2.5。
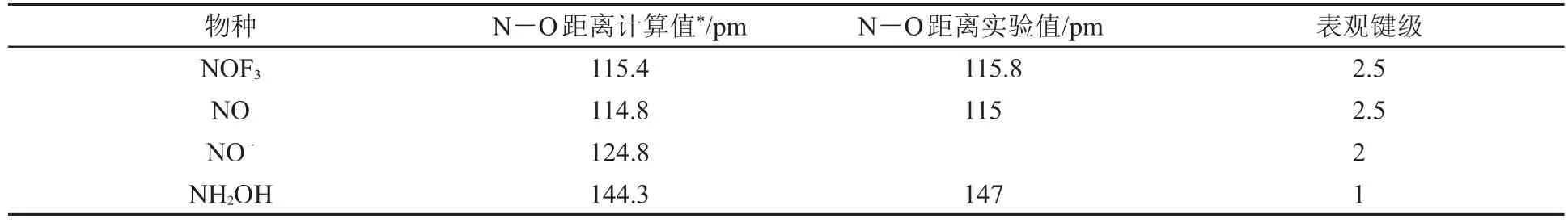
表2 不同物质N—O距离比较[14]
一种貌似聪明却不正确的解释是:Lewis结构中N带正电荷,O带负电荷,由于静电吸引作用使N—O距离缩短。须知:Lewis结构式上标的电荷是形式电荷。例如:NH4+的Lewis结构式中N带+1形式电荷,那么难道正电荷集中在电负性大的N上?这显然是不正确的,形式电荷不能作为判断实际电荷分布的依据。而且,这种说法也完全无法解释NOF3中N—F键变长的事实。(NF3中N—F键长137 pm,而NOF3中N—F键长143.1 pm,这违背了LCP模型[14]的预言。)如果换成POF3,可以用传统的d-p π键解释:“O的2p轨道上孤对电子进入P的3d轨道形成d-p π键”。但是现在的中心原子是氮,氮并没有可用的价层d轨道。
2.2p-π*相互作用
这里,我们提出一个新的理论来替代d-p π键。由群论或相关图可知,π反键轨道的对称性与d轨道相同。既然d轨道能和O的2p轨道对称性匹配,那么π反键轨道的对称性和O的2p轨道也匹配,所以O的2p轨道就能向NF3中(NOF3可以看成NF3+O)的π反键轨道提供电子,形成多重键——这就可以解释N—O缩短和N—F伸长的事实。
我们考虑O从很远处沿着NF3的C3轴不断接近NF3的过程。在O和NF3相距1.0 nm以上时,可以认为两者之间不存在相互作用。在接近过程中,我们人为地选取一些距离作为取样点,固定体系内N—O距离,并使体系强制保持C3v对称性(对称性限制条件),在单线态条件下进行优化。根据计算结果,我们绘制出了E-d(N—O)图(图3)和接近时的轨道图形变化情况(图4),以显示NF3中的π*轨道和O 的2p轨道之间的作用。(应当指出,NOF3为单重态,解离为NF3和自由O原子后(即上述拉近的逆过程),O为三重态,在上述过程中发生了多重度的变化,想要确切描述这一过程,严格来讲DFT方法是不够的,应该使用多参考态多组态方法。)
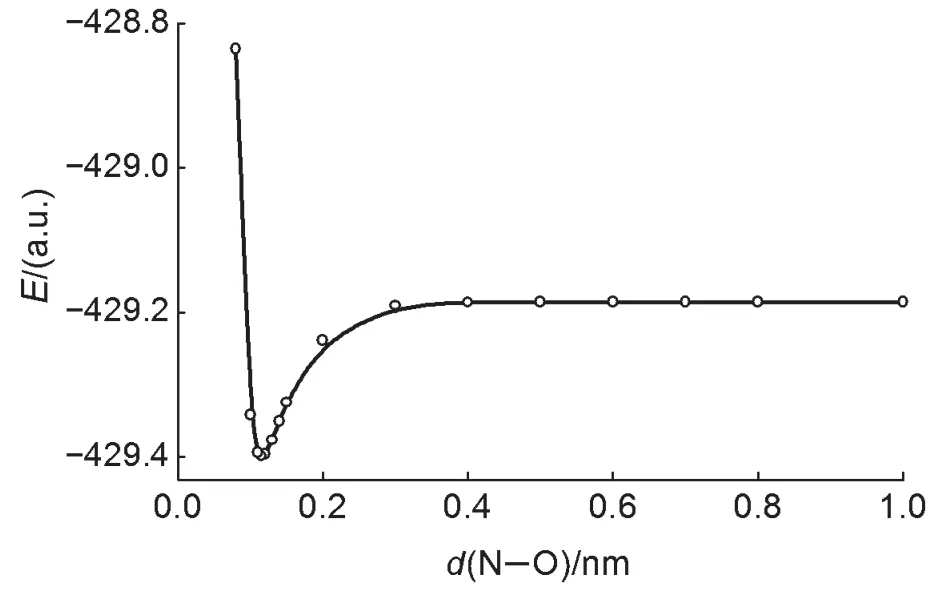
图3 O从C3轴接近NF3时的分子能量E随d(N—O)的变化曲线

图4 O从C3轴接近NF3时的轨道重叠图形变化情况
在O接近NF3的过程中,N—O距离达到0.4 nm前没有明显的相互作用,体系能量几乎不变。达到0.4 nm或更低时体系能量不断下降直至平衡点。N—O距离短于平衡距离后,体系能量迅速升高。在N—O距离达到0.2 nm时,p-π*的作用已经非常明显,如图4所示,O在逐渐接近NF3时,2p轨道逐渐与NF3的π*轨道发生叠加,并最终形成一条新的NOF3成键轨道和一条反键轨道。
需注意的是,NF3具有2个彼此正交的π*轨道(图5),而图4只绘制了其中一条的重叠情况。这2条轨道恰恰与2个O原子的2p轨道相匹配(2p轨道当然是正交的),因此会形成2个正交的简并成键轨道和2个正交的简并反键轨道。故在分子轨道理论水平上,d-p π键中的“d”不能理解为d轨道(真正动用d轨道形成π键的例子在过渡金属配合物中很常见,有π-给体或π-受体性质的配体同中心原子之间形成π键时,金属动用的就是d轨道。但是这个d轨道是内轨3d轨道(而非4d轨道),这种键一般也不被称为d-p π键。),而应理解为一条“有d轨道对称性的未占据分子轨道”。

图5 NF3中2条简并的π*轨道
另外,这种理论也解释了N—F键的伸长:反键轨道和p作用后填入电子,N—F键级减小,键长当然要伸长。至此,该理论很好地解释了NOF3中的种种“反常”。对于POF3,由于3d和2p能级不相近(至少没有3p和2p相近),所以仍应和NOF3一样用p-π*作用解释,而非用传统的“d-p π键”。
以上这种有趣的相互作用被Reed和Schleyer[15]解释为p-σ*的负超共轭作用。他们对NOF3的等电子体F3CO-进行了研究,认为O的p轨道与N—F σ*轨道发生作用。虽然这种解释可以较好地得到以上的分析结论,但是因为这个理论是基于“局部定域键”的,会导致O的p轨道由于空间取向,不可能同时与3条N—F σ*轨道发生作用。如果想解释3条N—F键等价,就还需要对氧的轨道再进行组合,这至少是不够方便的。
p-π*相互作用则不存在空间取向问题:如前面提到的,NF3有2条简并的π*轨道,它们彼此正交。这2条π*轨道恰好能和O的2条正交的p轨道叠加成键,不存在空间取向问题。所以我们认为离域的观点是更合适和直观的。
3 d轨道/d函数在超价分子中的作用
经过以上的分析和理论计算,我们认为d轨道在成键贡献中所占比例很有限。但是一些文献和理论计算表明:如果在计算中不加入d函数,将导致计算结果不收敛[16](计算结果不加d不收敛可能为当时的算法问题,现行Gaussian在计算中未发现此问题。),分子结构扭曲和键参数与实际情况差异较大。有人据此得出了d轨道在超价分子中不可或缺、起重要作用的结论。我们认为问题产生的根本原因在于混淆了d轨道和d函数的关系。
3.1d函数对超价分子计算结果的影响
为了说明d函数对分子的影响,我们选择了2个超价分子(SF6、SO42-)和1个非超价分子(SF2)进行了对比计算。分别采用含d函数和不含d函数的基组进行计算,所得结果列于表3。

表3 SO42--、SF6、SF2的计算和实验结果*
由表3和其他超价分子的分析可以得到以下结论:(1)从计算键长与实验键长比较可以看出,加入d函数可以改进计算精度,更接近真实值;(2)从能量降低可以看出,加入d函数后计算结果更接近“极小值”点,也更接近真实情况;(3)从SF6、SF2、SO42-的Δ键长结果看出,三者相差较为接近;(4)超价化合物SF6和不超价化合物SF2的ΔE/键数(平均每根键降低的能量)相近,在其他情况相同时,配位数高的化合物(一般为超价化合物)降低稍多。通过比较其他分子,还发现当配位原子为F、O时,键长变化更加明显。
3.2d函数与d轨道的关系
在计算化学中,一般将体系的波函数展开为一组基函数的线性组合,这一组基函数被称为基组(basic set)。体系波函数一般用原子的单电子波函数展开,称为Slater基组。若只取价电子轨道为基,则称为最小价基组。在计算中,最小价基组的局限性很大,需要加入更多的扩展基组来提高精度,给非H原子增加d函数,描述电子云的变形。例如p轨道变形拉长后,具有类似d轨道的性质,这些d函数(d function)称为d极化基组(d函数的名称来源于球谐函数,用来描述角向的空间分布,在这一点上与原子d轨道中d的含义是相同的,这就是二者的联系。)。通过加入极化基组提高计算结果的质量,是目前计算化学家广泛采用的技巧。但是,这样做仅仅是数学意义上的,就像在级数展开时,展开到更高阶可以更加逼近真实的函数。我们不能由加入d极化基组后计算结果的改善得到d轨道明显有成键作用的结论。
Magnusson[17]对d函数在第二、第三周期元素的超价化合物及正常化合物中的作用进行了分析,得到如下结论:在Hartree-Fock方法(HF)和微扰法(MP4)中,当d函数加到超价化合物及正常化合物中心原子的基组时,二者键能的增加程度没有显著差异,这与我们上面的分析结果一致。在HF法和微扰法中,d函数的具体作用不同,在HF方法中作为极化函数,在微扰法(MP4)中作为相关函数,但本质上都是为了提高计算的精度,克服Hartree-Fock方法中小基组的局限性。
此外,关于配体是O或F时键长优化效果明显的事实,我们认为可以这样解释:O和F具有较大的电负性和较多的孤对电子,这将导致中心原子周围具有较强的电场(尤其是在高配位时),这势必引起轨道的极化。因此,如果不加入极化基组,则和真实情况偏离较大,而加入后计算结果得到较大优化就是顺其自然的事了。
4 小结
纵观化学键理论的发展史,超价化合物是其中一个重要的节点,从Lewis结构理论开始一直到分子轨道理论,每种理论都试图提出自己的解释,而d轨道的作用成为其中一个有趣的话题。本文通过对超价化合物的几种解释和观点的分析,通过“经典”的和“非经典”的例子论述了外层d轨道杂化在描述超价分子时有关能量及贡献系数的不合理性,利用对称性和计算分析对d-p π键给出了p-π*作用的理解观点,最后阐述了计算化学中d函数与d轨道的关系,从而得出了传统的“d轨道参与”观点并不可靠的结论。
在现今的化学教学中,有很多教科书在讲述超价分子中的d轨道时,存在着误解或者避而不谈,我们认为这是很不合适的。至少应该让学生知道其中的问题所在,这样才能更深入地理解化学键的本质。
[1]Musher,J.I.Angew.Chem.Int.Edit.Engl.1969,8,54.
[2]徐佳,徐光宪,王祥云.化学通报,1986,No.3,46.
[3]Galbraith,J.M.J.Chem.Educ.2007,84,783.
[4]Forbus,T.R.;Martin,J.C.J.Am.Chem.Soc.1979,101(17),5057.
[5]Martin,J.C.Science 1983,221,509.
[6]Akiba,K.-y.;Yamashita,M.;Yamamoto,Y.;Nagase,S.J.Am.Chem.Soc.1999,121(45),10644.
[7]Yamashita M.;Yamamoto Y.;Akiba,K.-y.;Hashizume,D.;Iwasaki,F.;Takagi,N.;Nagase,S.J.Am.Chem.Soc.2005,127(12),4354.
[8]Kikuchi,Y.;Ishii,M.;Akiba,K.-y.;Nakai,H.Chem.Phys.Lett.2008,460,37.
[9]Pierrefixe,S.C.A.H.;van Stralen,S.J.M.;van Stralen,J.N.P.;Guerra,C.F.;Bickelhaupt,F.M.Angew.Chem.Int.Edit.Engl.2009,48(35),6469.
[10]Pierrefixe,S.C.A.H.;Guerra,C.F.;Bickelhaupt,F.M.Chem.Eur.J.2008,14(3),819.
[11]Pierrefixe,S.C.A.H.;Guerra,C.F.;Bickelhaupt,F.M.Chem.Eur.J.2008,14(23),6901.
[12]Greenwood,N.N.;Earnshaw,A.Chemistry of the Elements,1st ed.;曹廷礼,译.北京:高等教育出版社,1984.
[13]Frisch,M.J.;Trucks,G.W.;Schlegel,H.B.;et al.Gaussian 03,Revision C.02;Gaussian Inc.:Wallingford CT,2004.
[14]Gillespie,R.J.Corrd.Chem.Rev.2000,197,51.
[15]Reed,A.E.;Schleyer,P.V.R.J.Am.Chem.Soc.1990,112(4),1434.
[16]Magnusson,E.J.Am.Chem.Soc.1990,112(22),940.
[17]Magnusson,E.J.Am.Chem.Soc.1993,115(3),1051.
d Orbitals in Hypervalent Molecules
CHEN Tian-YangFAN Ru-BenCHEN Xiang-YuCUI Zhi-Hao*
(College of Chemistry and Molecular Engineering,Peking University,Beijing 100871,P.R.China)
Based on the Gaussian DFT calculations and analysis on hypervalent molecules,we concluded that the theory that outer valence d orbitals undergo hybridization and get involved in the formation of the d-p π bond is not reasonable.We proposed an explanation that can interpret experimental facts.Additionally,we described the relationship between the d orbital and the d function,which are not equal in computational chemistry.
Hypervalent molecules;d orbital;Hybrid orbital theory;d-p π bond;Polarization basis sets
O6;G64
10.3866/PKU.DXHX20160238
,Email:zhcui@pku.edu.cn
猜你喜欢
高中数理化(2023年6期)2023-08-26 13:28:24
辽宁科技大学学报(2022年5期)2023-01-04 12:45:34
原子与分子物理学报(2020年5期)2020-03-17 06:59:34
火工品(2019年6期)2019-06-05 02:35:44
陕西中医(2018年6期)2018-08-29 00:43:34
考试周刊(2018年39期)2018-04-19 10:39:44
厦门大学学报(自然科学版)(2018年2期)2018-04-11 07:07:35
中国塑料(2016年1期)2016-05-17 06:13:00
读写算·教研版(2016年8期)2016-05-07 11:52:08
中国塑料(2016年11期)2016-04-16 05:25:55
