
PO2X…PX3/PH2X(X=F,Cl,Br,CH3,NH2)复合物中π-ho le磷键作用的电子密度拓扑分析
2016-09-13 03:10:20王月红李晓艳曾艳丽孟令鹏张雪英
物理化学学报 2016年3期
王月红 李晓艳 曾艳丽 孟令鹏 张雪英
(河北师范大学化学与材料科学学院,河北省无机纳米材料重点实验室,石家庄050024)
PO2X…PX3/PH2X(X=F,Cl,Br,CH3,NH2)复合物中π-ho le磷键作用的电子密度拓扑分析
王月红李晓艳曾艳丽孟令鹏张雪英*
(河北师范大学化学与材料科学学院,河北省无机纳米材料重点实验室,石家庄050024)
磷键作为一种新型的分子键合力,因在晶体工程和超分子合成等方面的重要作用而越来越多地引起科研工作者的广泛关注。本文采用量子化学从头算和电子密度拓扑分析等方法,在MP2/aug-cc-pVTZ理论水平上,对PO2X…PX3和PO2X…PH2X(X=F,C l,Br,CH3,NH2)π型复合物的结构和磷键性质进行了理论研究。研究表明:π-hole磷键复合物存在A和B两种稳定构型,分别以P…P和P…X磷键作用为主。分子中原子(AIM)、非共价作用(NCI)、电子定域函数(ELF)及适应性自然密度划分(AdNDP)分析表明,不同取代基对该类磷键作用的性质产生很大影响:当取代基为给电子基(CH3,NH2)时,磷键具有明显的共价作用特征;当取代基为吸电子基(F,Cl,Br)时,构型和取代基不同的磷键分别表现为非共价、部分共价或共价作用特征。自然键轨道(NBO)分析指出,分子间磷键的Wiberg键级的数值越大,磷键的共价性越强,磷键的作用强度越大。构型B的电荷转移主要是PX3/PH2X中X原子上的孤对电子转移到PO2X中π*(P=O)反键空轨道。
分子间相互作用;磷键;π-hole作用;NCI分析;电子密度拓扑分析;ELF分析
1 引言
非共价相互作用在超分子化学、分子生物学、晶体工程、材料自组装等领域发挥着非常重要的作用1,2。氢键和卤键作为重要的非共价相互作用,一直以来是理论和实验科研工作者的研究热点3,4,近几年,科研工作者发现了一种新型的非共价相互作用——磷键。磷键也是一种路易斯酸和路易斯碱之间的相互作用,其中磷族原子(N,P,As)充当路易斯酸5。磷键作为一种新的分子键合力,对物质多层次构筑具有重要意义6。
早在几十年前人们就知道磷族原子(N,P,As)可以作为路易斯酸7,但直到2011年Scheiner等8-10开始了磷键作用的研究,他们在蛋白质及一些晶体结构中发现了P…N作用,并确认是一种新的非共价相互作用。随后Hey-Hawkins等6的工作引起了科学界对磷键的广泛关注,自此,科研工作者对这种非共价作用的结构及性质进行了大量深入的研究8-29。Scheiner等11-14对一系列P…D类型的(其中P表示PH3及一些膦类化合物;D表示含N、O、S等给电子基的分子以及乙烯、乙炔和苯环等)磷键复合物进行了研究,指出磷键与氢键、卤键的共同点及区别。Del Bene等15-18研究了多种复合物(PH2X)2,H2XP:NXH2,(PH2X)3和(PHFX)2中的P…P和P…N磷键,以及sp2杂化的磷原子化合物组成的磷键19-22。Li等23-25讨论了取代、协同及溶剂效应对磷键的影响,分析了磷键和卤键、阳离子…π作用之间的协同效应等。另外科研工作者还对一些特殊的磷键如阴离子磷键26、阳离子磷键27、磷氢键28和单电子磷键29等进行了预测和分析。
在解释卤键的形成原因时,Clark等30提出了“σ-hole”的概念,用于描述在R―X共价键伸展方向上卤原子外层中心的静电势为正值的区域,而其余的大部分区域静电势为负值。随后,人们常用“σ-hole”的概念来预测和解释IV-VIIA主族原子参与的分子间相互作用31,32。随着对分子间相互作用研究的深入,Murray等33又定义了与“σhole”相似的“π-hole”,即在垂直于分子骨架的区域存在的正静电势区。正的“σ/π-hole”可以与负电荷的浓集区(如阴离子、路易斯碱的孤对电子或π电子)发生相互作用,具有高度的方向性。对σ/πhole作用的研究也是近年来的热点课题。Del Bene等19重点讨论了复合物H2C=(X)P…PXH2(X=F, Cl,OH,CN,NC,CCH,H,CH3,BH2)中涉及σ-hole和π-hole的P…P磷键,Solimannejad等34,35分别对SO2及O2NX(X=Cl,Br,I)中的π-hole、σ-hole及相互作用进行了理论探讨。Frontera等36研究了YO2Br (Y=N,P,As),CF3NO2,CH3NO2与富电子的分子之间的π-hole磷键作用。曾艳丽等37分析了ClO…XONO2/XONO…NH3(X=Cl,Br,I)体系中σ-hole和π-hole的协同作用。σ/π-hole作用在晶体组装中起着至关重要的作用,对固态物质的结构产生很大影响,具有广阔的应用前景38。PO2Cl是一种典型的含π-hole的磷化合物,Brupbacher-Gatehouse39首次对气相PO2Cl分子进行了光谱研究,明确了分子构型和电子结构,Del Bene等40分析了PO2X(X= F,Cl)中的π-hole与含N的路易斯碱之间的相互作用。本文在前人工作的基础上,为了明确不同类型取代基对分子间磷键性质的影响,探讨π-hole磷键的作用本质,选择PO2X与PX3、PH2X(X=F, Cl,Br,CH3,NH2)为研究体系,依据分子中原子的量子理论(QTAIM)和电子定域函数(ELF)等理论,采用量子化学计算方法进行研究。文章从复合物的构型、相互作用能、分子间的电荷转移性质、键鞍点处的电子密度和能量密度性质、电子定域函数以及非共价作用指标等方面对π-hole磷键作用进行较全面的讨论和分析。
2 计算方法
研究表明应用二级微扰理论(MP2)结合中等大小的基组(如aug-cc-pVTZ),对氢键、卤键、磷键等分子间相互作用的研究可以得到合理的键能及复合物稳定构型等可靠的数据18,21,24,因此本文的计算均在MP2/aug-cc-pVTZ计算水平上进行。对所有单体和复合物的构型优化及同一水平上的频率计算,证明优化所得的稳定构型是这个势能面上的最小点。分子间相互作用能是用复合物的能量减去单体能量之和,其中能量进行了基组重叠误差41和零点能校正。以上计算均使用Gaussian 09程序包42完成。
QTAIM指出43-45,键鞍点处的电子密度和能量密度与化学键的性质有着密切的联系。当键鞍点处的电子密度Laplacian量(▽2ρb)小于零时,键鞍点处电荷密集、化学键具有共价键特征;当▽2ρb>0时,键鞍点处电荷发散,一般为闭壳层相互作用如离子键、氢键和范德华相互作用。如果▽2ρb为正值,电子能量密度(Hb)为负值,则该相互作用具有部分共价性质。动能密度(Gb)和势能密度(Vb)之间的相对大小决定了相互作用的本质:如果-Gb/Vb大于1,该相互作用为非共价性质,以静电作用成分为主;如果-Gb/Vb介于0.5和1之间,则为部分共价性质,数值越大,静电作用成分更大些;如果-Gb/Vb小于0.5,具有共价性质,数值越小,共价作用特征越强。ELF理论指出46-49,复合物分子中两原子间的一维ELF曲线的极大值反映了原子间相互作用的性质,若间隙区域ELF值很低,两原子间为静电作用;若连接两原子间的ELF极大值在0.6至1.0之间,则为共价作用。
本文依据QTAIM和ELF理论进一步探讨πhole磷键的作用本质,在优化构型的基础上,使用WFA程序50计算得到单体分子的静电势;使用AIM2000程序包51进行电子密度拓扑分析;采用Multiw fn程序包52完成ELF、非共价作用(NCI)及适应性自然密度划分(AdNDP)分析计算,通过VMD软件53可视化输出NCI及AdNDP分析计算结果。自然键轨道分析(NBO)是采用Gaussian09程序包中内置的NBO程序完成的,NBO轨道图采用Multiw fn程序和VMD程序绘制。
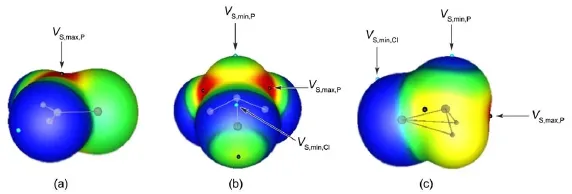
图1 分子(a)PO2Cl,(b)PCl3和(c)PH2Cl在电子密度为0.001 a.u.等值面上的静电势图Fig.1 Electrostatic potentialson themolecu lar surfacesof(a)PO2Cl,(b)PCl3,and(c)PH2Clmolecu les, com pu ted on the 0.001 a.u.contour of the electronic density VS,max:localmostpositiveelectrostatic potentials;VS,min:localmostnegative electrostatic potentials
3 结果与讨论
3.1单体分子的静电势分析
分子静电势54,55是一种很好的研究非共价作用的工具,可以很好地预测和分析包括氢键、卤键等分子间相互作用的方向和强度31,34,37,38。图1以PO2Cl,PCl3和PH2Cl为代表给出了在电子密度为0.001 a.u.(electrons∙Bohr-3)的等值面上的分子静电势图。可以看出在PO2Cl分子的上方和下方,存在着静电势为正值的红色区域,对应π-hole,VS,max表示静电势的极大值;氧原子周围为蓝色的负静电势区,对应氧原子上的孤对电子。对于PCl3分子来说,沿着P―Cl共价键延长线上的P端存在三个正静电势的红色区域即σ-hole,氯原子周围为负静电势区,VS,min表示局部静电势极小值;磷原子的顶端也存在局部静电势最小区,对应磷原子上的孤对电子。对于PH2Cl分子来说,沿着P―Cl共价键延长线上的P端也存在σ-hole,氯原子周围和磷原子的顶端呈现蓝色的负静电势区域。通过静电势分析可以预测,PO2X分子中的π-hole可以作为电子受体与PX3和PH2X中P原子或X原子附近的负静电势区作用,形成π-hole磷键;PX3和PH2X分子中的σ-hole可以作为电子受体和PO2X中O原子周围的负静电势区作用,形成σ-hole磷键。
表1给出了各单体分子表面的静电势数值。可以看出,PO2X中π-hole的静电势极大值VS,max,P的数值大于PX3和PH2X中σ-hole的VS,max,P数值。可以预测,以π-hole作用形成的磷键要强于以σ-hole作用形成的磷键。从表1数据还可看出,σ/π-hole的VS,max,P值随取代基X的不同而变化,吸电子基(F, Cl,Br)的VS,max,P数值大于给电子基(CH3,NH2)的VS,max,P值,随着F、Cl、Br的电负性减小,VS,max,P数值逐渐减小。从PX3和PH2X分子中静电势的极小值VS,min,P可以看出,给电子取代基使得P原子周围的电子密度增加,VS,min,P数值更负。当取代基为吸电子基时,由于卤原子的吸电子能力使得PX3中P原子上孤对电子所对应区域的VS,min,P变为正值。比较PX3和PH2X分子中卤素取代基所对应的静电势极小值,发现单取代分子的VS,min,P和VS,min,X值均比三取代分子相应的数值要负。
3.2复合物的几何构型和相互作用能
为了区别不同化合物中的磷原子,以Pd表示PO2X中的P原子,Ps表示PX3和PH2X中的P原子。虽然XO2P…PX3和XO2P…PH2X复合物的分子势能面上存在多个极小点,但本文限定研究范围为磷键复合物的结构及性质。首先以XO2P…PH2X (X=F,Cl,Br,CH3,NH2)为例,比较以π-hole和σhole作用形成的磷键复合物的稳定性。在MP2/ aug-cc-pVTZ理论水平上的计算发现,以PO2X中的π-hole为电子受体形成的磷键复合物比以PX3和PH2X中σ-hole为电子受体形成的磷键复合物稳定得多。例如XO2P…PH2X(X=F,Cl,Br,CH3, NH2)之间π-hole磷键作用的相互作用能分别为-72.5、-47.6、-42.3、-50.5、-51.5kJ∙mol-1(见表2),而σ-hole磷键作用的相互作用能分别为-8.5、-12.5,-13.1,-13.7,-23.9 kJ∙mol-1(构型见图S1,见Supporting Information),σ-hole磷键作用能远小于相应分子间π-hole作用的相互作用能。因此稳定的磷键复合物以π-hole作用为主,这与前面的静电势分析一致,因此本文主要讨论以PO2X中的π-hole为电子受体形成的磷键复合物的结构和性质。

表1 分子PO2X、PX3和PH2X(X=F,Cl,Br,CH3,NH2)静电势的局部极大值(VS,max)和极小值(VS,min)Tab le1 Calculated VS,maxon Patom and VS,minassociated with the Pand X atomsof PO2X, PX3,and PH2X(X=F,Cl,Br,CH3,NH2)monomers
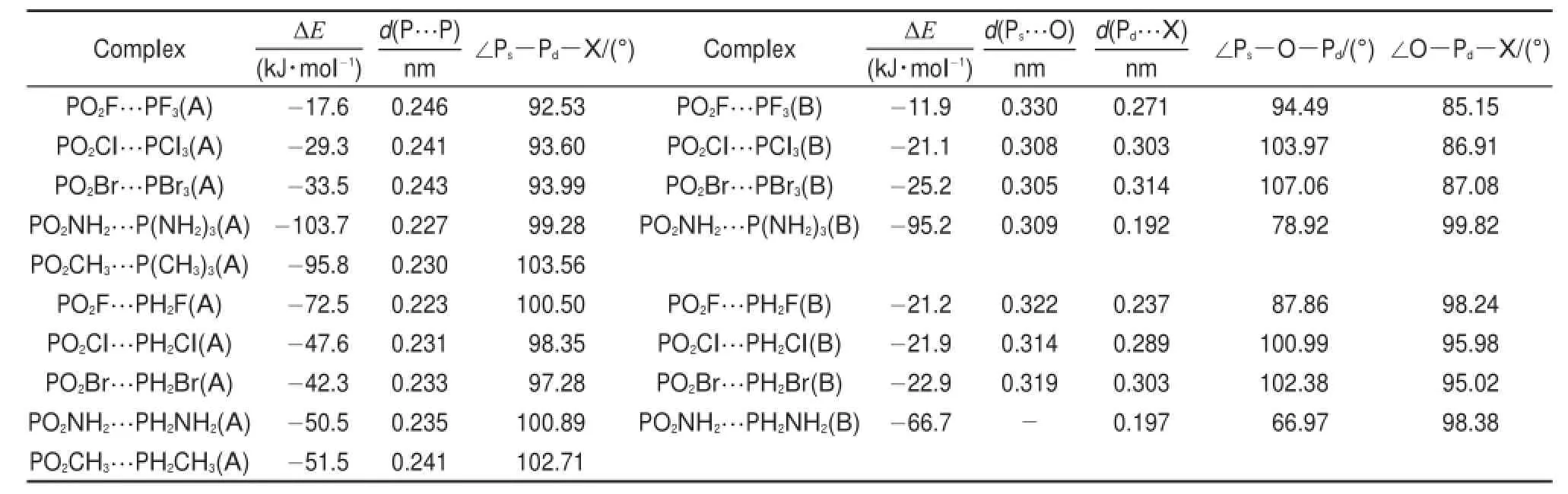
表2 π-hole磷键复合物的主要构型参数和相互作用能Table2 Main geometricalparametersand interaction energiesof theπ-hole pnicogen-bonded com plexes
表2给出了π-hole磷键复合物中几个重要的构型参数和分子间相互作用能。图2以PO2Cl…PCl3、PO2NH2…P(NH2)3和PO2Cl…PH2Cl、PO2NH2…PH2NH2为代表,给出了π-hole磷键复合物的两种稳定构型。以PO2X中的π-hole为电子受体可形成两种磷键复合物结构,一种是PX3和PH2X中的Ps为电子给体与π-hole作用,标为A构型,另一种是PX3和PH2X中的X为电子给体与π-hole作用,标为B构型(P(CH3)3和PH2CH3除外)。从表2中数据可以看出,X为不同类型的取代基时,相互作用能发生较大变化。例如PO2X…PX3构型A复合物,当X=F, Cl,Br时,相互作用能分别为-17.6、-29.3、-33.5kJ∙mol-1,而当X=NH2,CH3时,相互作用能大得多,分别为-103.7和-95.8 kJ∙mol-1。这可以从分子静电势的分析看出:由于卤原子的吸电子作用,使得PX3中P原子上的静电势极小值为正值,因此PX3与PO2X(X=F,Cl,Br)之间的A构型复合物相互作用能较小。给电子基(NH2,CH3)大大增加了PX3中P原子上的电子密度,使得P原子上的静电势极小值VS,min由PF3的7.57 k J∙mo l-1变到P(CH3)3的-114.36kJ∙mol-1,从而分子间相互作用能由-17.6kJ∙mol-1大大增加到-95.8 kJ∙mol-1。对NH2取代基,体系中还存在着N…H和O…H氢键作用,因此相互作用能很大。从表2还可看出,当PX3和PH2X中的X为单取代和多取代时,相互作用能也不同。PO2X…PX3/PH2X间形成A构型复合物时,卤素三取代形成的复合物不如单取代形成的复合物稳定,而CH3、NH2等给电子基的三取代复合物所对应的相互作用强度大于单取代复合物对应的作用强度,这也与PX3和PH2X中Ps原子附近的静电势极小值成正相关,静电势越负,相互作用能越负,相互作用的强度越大。
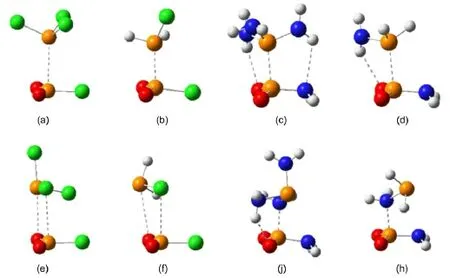
图2 复合物PO2Cl…PCl3,PO2NH2…P(NH2)3和PO2Cl…PH2Cl,PO2NH2…PH2NH2构型A和构型B的稳定构型图Fig.2 Op tim ized geometriesof the rep resentative con formation A and B comp lexes PO2Cl…PCl3, PO2NH2…P(NH2)3,PO2Cl…PH2Cl,and PO2NH2…PH2NH2(a)PO2Cl…PCl3(A);(b)PO2Cl…PH2Cl(A);(c)PO2NH2…P(NH2)3(A);(d)PO2NH2…PH2NH2(A);(e)PO2Cl…PCl3(B); (f)PO2Cl…PH2Cl(B);(j)PO2NH2…P(NH2)3(B);(h)PO2NH2…PH2NH2(B)
3.3电子密度及能量密度性质分析
分子中原子的量子理论(QTAIM)43,44将有关化学概念如化学结构、化学键、化学反应性质等与电子密度分布函数的拓扑性质联系起来,定量化地直观描述了分子中原子及原子间化学键,电子密度拓扑分析是一种研究非共价作用的有效手段56,57。图3给出了以PO2Cl…PCl3、PO2NH2…P(NH2)3和PO2Cl…PH2Cl、PO2NH2…PH2NH2构型A和构型B为代表的AIM分子图。图中Pd…Ps、Ps…O和Pd…Cl、Pd…N原子之间的键鞍点及键径证实了分子间磷键作用的存在。表3给出了复合物中分子间相应键鞍点处的电子密度和能量密度数据:电子密度(ρb)及其Laplacian量(▽2ρb)、动能密度(Gb)、势能密度(Vb)、总的电子能量密度(Hb)及比值(-Gb/Vb)。
从表3数据可以看出,对于构型A复合物,无论取代基为吸电子基或给电子基,所有Pd…Ps间键鞍点处的▽2ρb值小于零,Hb值小于零,-Gb/Vb值小于0.5,说明它们间的磷键为共价作用。对于构型B复合物,当取代基X为给电子基(NH2)时,复合物中Pd…N键鞍点处▽2ρb接近于零,Hb<0,-Gb/Vb≈0.5,这类分子间作用也具有明显的共价作用特征;当取代基X为吸电子基(F,Cl,Br)时,除了PO2F…PF3(B)外,其他复合物中Pd…X间键鞍点处的▽2ρb为正值,Hb值小于零,-Gb/Vb值在0.8左右,说明分子间磷键具有部分共价作用性质,静电作用的成分大一些;PO2F…PF3(B)复合物中Pd…F键鞍点处▽2ρb>0,Hb>0,-Gb/Vb>1,分子间磷键为非共价作用,以静电作用为主。
3.4非共价相互作用分析
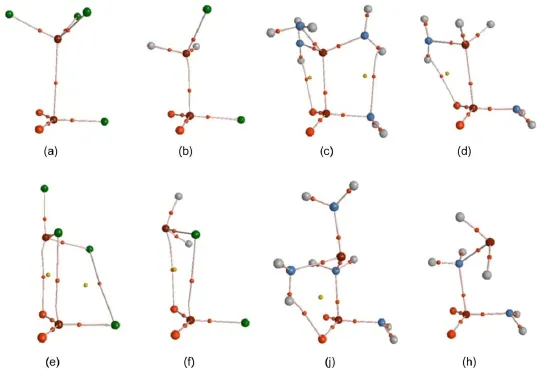
图3 复合物PO2Cl…PCl3,PO2NH2…P(NH2)3,PO2Cl…PH2Cl和PO2NH2…PH2NH2构型A和构型B的分子图Fig.3 Molecular graphsof the rep resentative con formation A and B comp lexes PO2Cl…PCl3, PO2NH2…P(NH2)3,PO2Cl…PH2Cl,and PO2NH2…PH2NH2(a)PO2Cl…PCl3(A);(b)PO2Cl…PH2Cl(A);(c)PO2NH2…P(NH2)3(A);(d)PO2NH2…PH2NH2(A);(e)PO2Cl…PCl3(B); (f)PO2Cl…PH2Cl(B);(j)PO2NH2…P(NH2)3(B);(h)PO2NH2…PH2NH2(B)
非共价作用(NCI)分析方法是由杨伟涛课题组58,59基于电子密度和约化密度梯度函数(RDG,the reduced density gradient)的分析提出的一种新的分析方法,该方法能够形象地表示出非共价相互作用的类型和强度,不仅可用于氢键、卤键、静电作用、范德华作用等弱相互作用的研究,还能展现出强互斥作用(例如环和笼中的位阻效应)。NCI分析方法在某种程度上可以视为QTAIM方法的扩展。该方法指出,如果在关键点处ρb>0,λ2为负值,即sign(λ2)=-1,则相互作用表现为强吸引作用(例如偶极-偶极作用或氢键);如果关键点处λ2为正值,即sign(λ2)=+1,sign(λ2)ρb为较大的正值,则相互作用表现为强互斥作用(例如环和笼中的位阻效应);如果sign(λ2)ρb≈0,那么相互作用表现为范德华作用。若将ρb和λ2的符号相乘而得的sign(λ2)ρb函数用不同色彩投影到RDG等值面上,则相互作用的位置、强度、类型都能一目了然地显现出来。当sign(λ2)=-1,且ρb在低密度区(0.005a.u.<ρb<0.05a.u.)时,体系中若存在分子间相互作用,则在RDG对应sign(λ2)ρb的散点图中将出现垂直于sign(λ2)ρb数值的“峰钉”,且ρb数值越大,相互作用的强度越大55,56。RDG填色等值面图根据sign(λ2)ρb数值大小显示不同的颜色,按照蓝-绿-红的趋势逐渐变化,其中蓝色越深表示相互作用越强,越接近红色表示排斥作用越大。
本文以PO2Cl…PCl3(A)、PO2Cl…PH2Cl(A)和PO2Cl…PH2Cl(B)为例,给出了RDG对应sign(λ2)ρb的散点图和RDG填色等值面图(如图4所示)。从图4(a)和4(b)中的散点图可以看到,其sign(λ2)ρb数值分别为-0.080和-0.097 a.u.,其RDG填色等值面图中出现蓝色区域,说明PO2Cl…PCl3(A)和PO2Cl…PH2Cl(A)复合物分子间作用强度较大,以共价成分为主,这和前面QTAIM分析是吻合的。复合物PO2Cl…PH2Cl(B)中(图4(c)),在sign(λ2)ρb数值分别为-0.024和-0.009 a.u.处出现“峰钉”,分别对应于Pd…Cl和O…Ps间的非共价相互作用;填色等值面图显示了Pd…Cl和O…Ps之间的成键等值面分别为蓝绿色和绿色。从sign(λ2)ρb数值和成键颜色可看出Pd…Cl间的作用强度大于O…Ps间的作用强度,这和前面的QTAIM分析也是一致的。对所有磷键复合物的NCI分析发现,当sign(λ2)ρb数值大于-0.05a.u.时,散点图和RDG填色等值面图与PO2Cl…PH2Cl(A)相似,分子间作用以共价成分为主,这些体系分别为取代基为CH3、NH2的复合物和取代基为卤素的构型A复合物,此结果与QTAIM分析的结论一致。取代基为卤素的构型B复合物的NCI分析散点图及RDG填色等值面图与PO2Cl…PH2Cl(B)相似,磷键作用表现为非共价成分为主。

表3 π-hole磷键复合物中键鞍点处的电子密度和能量密度性质Table3 Electron and energy densitiesat thebond criticalpointsofπ-hole pnicogen-bonded comp lexes
3.5电子定域函数分析
电子定域函数(ELF)理论是由Becke和Edgecombe46提出的,随后Savin等47,48发展和应用了ELF理论。利用ELF理论方法,可以分析原子的壳层结构,确定分子中孤对电子浓集点的方向和位置,研究分子的化学键结构、反应过程化学键的变化等49。
图5(a)和5(b)给出了复合物PO2Cl…PCl3(A)、PO2Cl…PH2Cl(A)沿着Pd…Ps间作用方向的一维ELF函数曲线和二维ELF函数等值面图,图5(c)是PO2Cl…PH2Cl(B)沿着Pd…Cl间作用方向的关系图。从一维函数曲线可以看出,在PO2Cl…PCl3(A)和PO2Cl…PH2Cl(A)中(图5(a)和5(b)),两个磷原子的壳层结构发生明显的变化,两磷原子间隙区域的ELF极大值分别为0.96和0.95,说明两分子间发生了较强的相互作用,磷键属于共价作用。在PO2Cl…PH2Cl(B)中(图5(c)),两个磷原子的壳层结构在形成复合物前后没有明显的变化,说明PO2Cl…PH2Cl(B)分子间属于非共价作用,对磷原子的壳层结构影响不大。从ELF的二维等值面图也可以看出分子中孤对电子(红色区域)的方向和位置。在复合物PO2Cl…PCl3(A)和PO2Cl…PH2Cl(A)中(图5(a)和5(b)),PCl3和PH2Cl中P的孤对电子对应着PO2Cl中的P原子上的电子发散区域,即πhole;在复合物PO2Cl…PH2Cl(B)中(图4(c)),PH2Cl中Cl上的孤对电子对应着PO2Cl中的πhole,PO2Cl中O上的孤对电子与PH2Cl中Ps的σ-hole相对应。值得注意的是,复合物PO2Cl…PCl3(A)和PO2Cl…PH2Cl(A)中两个磷原子之间出现了键域,说明两个磷原子之间有明显的共价作用,而在复合物PO2Cl…PH2Cl(B)中,磷原子和氯原子之间没有键域,表明分子间的共价成份较低。对所有复合物的ELF分析发现,对于取代基为CH3、NH2的复合物和取代基为卤素的构型A复合物,分子间均存在相互作用的键域,相互作用原子间的ELF极大值均在0.91以上,说明这些分子间磷键具有明显的共价作用特征。对于取代基为卤素的构型B复合物,其ELF一维函数曲线和二维等值面图与PO2Cl…PH2Cl(B)相似,分子间磷键的共价成分较低,这和前面AIM和NCI分析的结果一致。
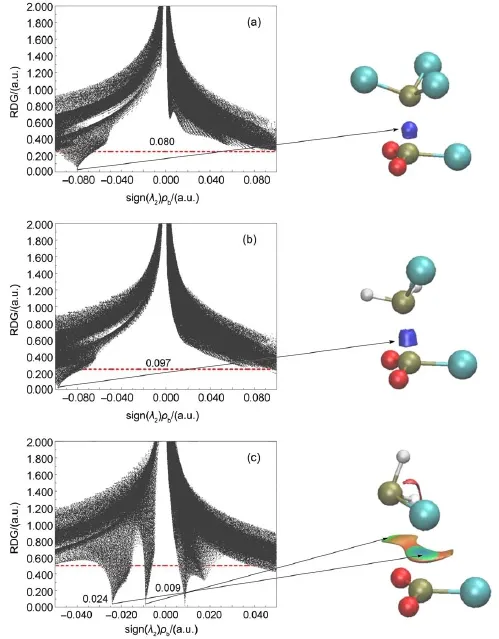
图4 在MP2/aug-cc-pVTZ水平上磷键复合物的散点图(左)和填色RDG等值面图(右)Fig.4Scatter d iagram(left)and color-filled RDG isosurfacem ap(right)of pnicogen-bonded com p lexesat theMP2/aug-cc-pVTZ level (a)PO2Cl…PCl3(A),(b)PO2Cl…PH2Cl(A),(c)PO2Cl…PH2Cl(B);RDG:the reduced density gradient
3.6自然键轨道分析
自然键轨道(NBO)分析强调轨道作用,可以从轨道相互作用的角度来理解复合物的形成过程,可以定量衡量电荷供体和受体间的电荷转移量。为了研究磷键作用的本质,在HF/aug-cc-pVTZ水平上对所研究的体系进行了NBO分析。NBO方法对相互作用能大于-71 kJ∙mol-1的分子间作用不能给出很好的分析结果40,研究发现此方法也不适用于具有明显共价作用特征的分子间相互作用体系。根据前面的QTAIM和NCI分析结果,表4给出了具有非共价及部分共价作用特征的π-hole磷键的NBO分析结果,其中E(2)为电荷供体及受体轨道间相互作用的二级稳定化能,Δq为供体受体间的电荷转移数,可以通过以下公式计算得到:
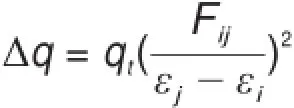
其中qi是轨道占据数,εi、εj是Fock矩阵对角元素,Fij为Fock矩阵非对角元素。
从表4可以看出,除了PO2F…PF3(B)复合物外,构型B复合物分子间的电荷转移包含两部分,一部分是PX3/PH2X中X原子上的孤对电子到PO2X中P=O反键轨道的电荷转移(LP(X)→BD*(P=O)),另一部分是PO2X中O原子上的孤对电子到PX3/PH2X中P―X/P―H反键轨道的电荷转移(LP (O)→BD*(P―X/P―H)),其中第一种电荷转移为主,占电荷转移总量的80%以上,其轨道间相互作用的强度(E(2))也远大于第二种电荷转移,此结论与QTAIM、NCI分析得到的结论一致:Pd…X间的作用强度大于O…Ps间的作用强度。图S2(见Supporting Information)给出了两个代表性复合物PO2Cl…PCl3(B)和PO2Cl…PH2Cl(B)的NBO轨道图,图S2(a)和S2(b)分别给出了PO2Cl…PCl3(B)中的LP(Cl)→BD*(P=O)和LP(O)→BD*(P―Cl)轨道作用图,图S2(c)和S2(d)分别给出了PO2Cl…PH2Cl (B)中的LP(Cl)→BD*(P=O)和LP(O)→BD*(P―H)轨道作用图。从图中可以清楚地看出电荷给体和受体轨道之间的作用,PO2Cl分子中P=O反键轨道为π轨道,即π-hole;PCl3/PH2Cl中P―Cl和P―H反键轨道为σ轨道,即σ-hole。因此PO2Cl…PCl3(B)和PO2Cl…PH2Cl(B)分子间作用为σ-π和σ-σ型磷键,从E(2)和Δq的数值大小可看出σ-π型磷键作用强度更大。
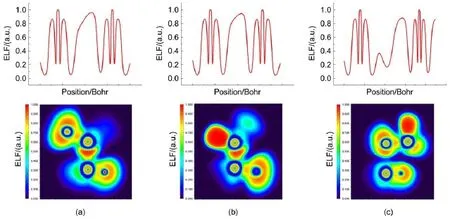
图5 磷键复合物的ELF的一维函数曲线图(上)和二维等值面图(下)Fig.5One dimensional(up)and two dimensional(down)mapsof ELFof pnicogen-bonded comp lexes (a)PO2Cl…PCl3(A),(b)PO2Cl…PH2Cl(A),(c)PO2Cl…PH2Cl(B);FLF:electronic localized function
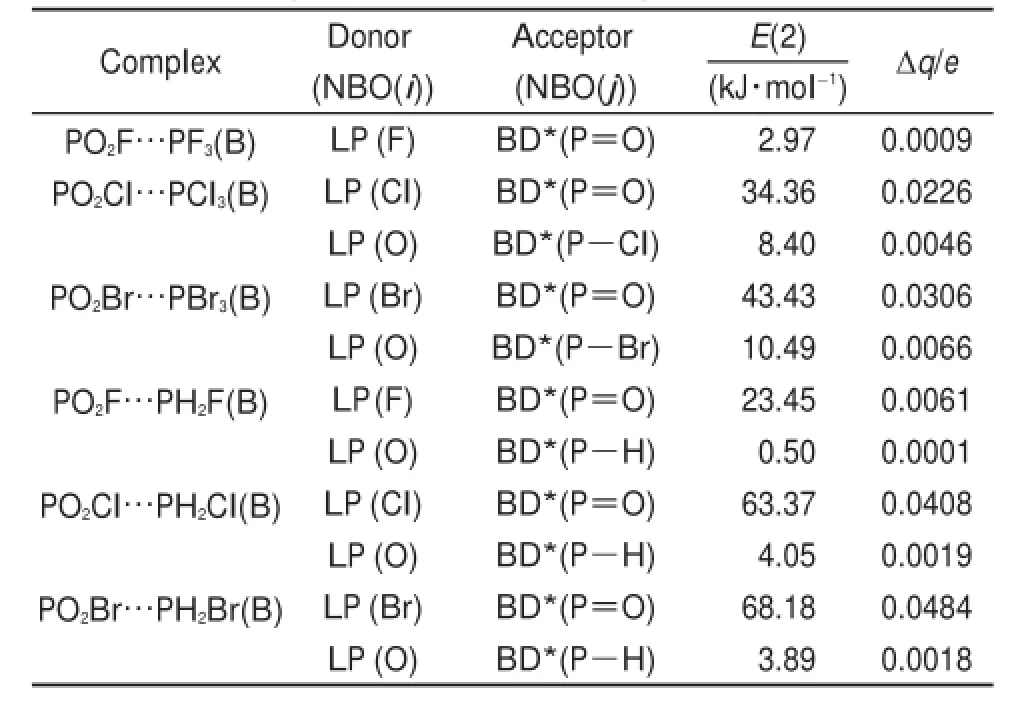
表4 π-hole磷键复合物的自然键轨道分析Tab le 4Naturalbond orbitalanalysisofπ-hole pnicogen-bonded com plexes
分析表4中的数据,发现卤素三取代PX3复合物的二级稳定化能E(2)和电荷转移量Δq均小于卤素单取代PH2X复合物的相应数值,电荷给体与受体间轨道相互作用的强度以及电荷转移量均与磷键的相互作用能大小顺序一致。例如,PO2F…PF3(B)、PO2Cl…PCl3(B)、PO2Br…PBr3(B)的E(2)值分别为2.97、34.36和43.43 kJ∙mol-1,从卤原子的孤对电子到P=O反键轨道的电荷转移量分别为0.0009e、0.0226e和0.0306e,它们的相互作用能分别为-11.9、-21.1和-25.2 kJ∙mol-1。电荷给体和受体轨道间的相互作用越大,电荷转移越多,磷键的相互作用强度越大,表明轨道间的相互作用在磷键体系中也起着重要的作用。
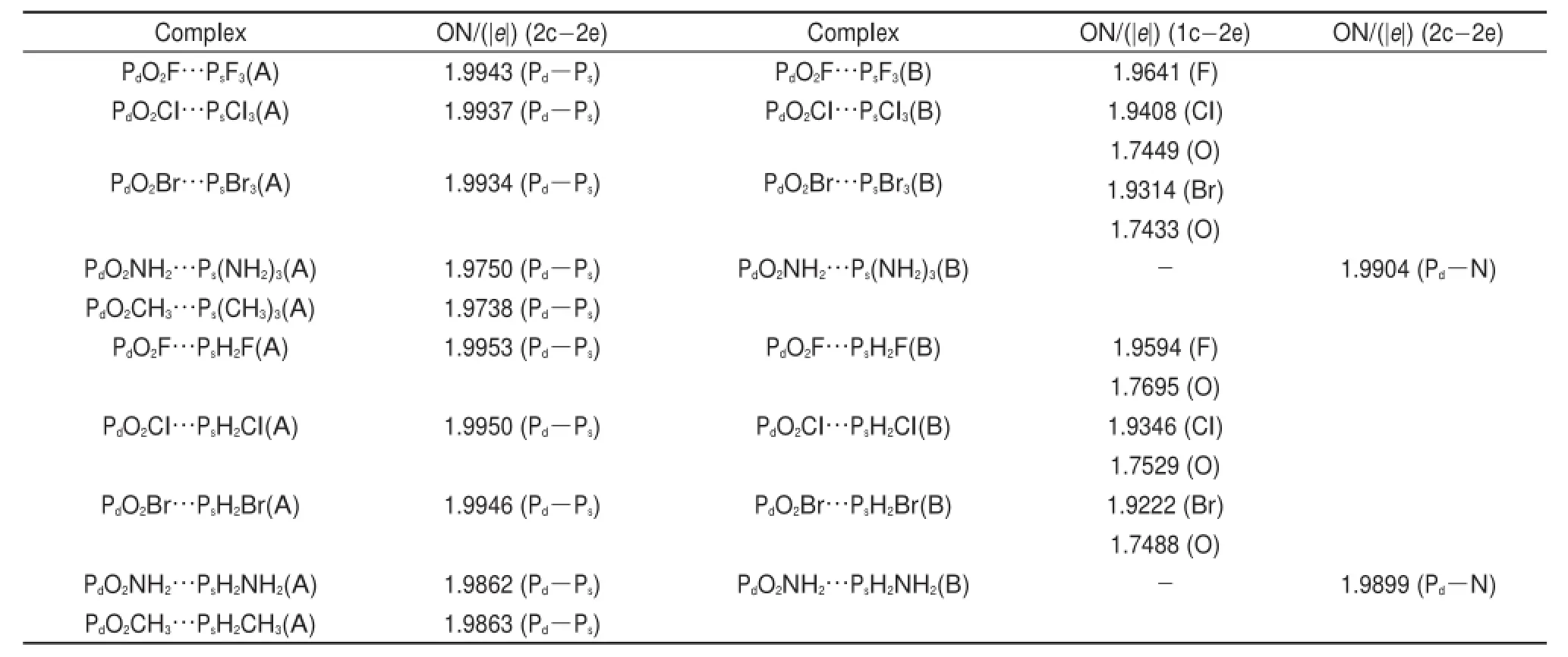
表5 π-hole磷键复合物的适应性自然密度划分方法分析Tab le 5Adaptive natural density partitioning analysisofπ-hole pnicogen-bonded com p lexes
表S1(Supporting Information)给出了磷键复合物相应键鞍点处的W iberg键级数据,从表S1的数据可知,磷键的Wiberg键级的大小顺序和相互作用能以及键鞍点处的电子密度的变化顺序是一致的,即Wiberg键级的数值越大,键的强度越大。并且Wiberg键级的数值越大,键的共价性越强,这与AIM及NCI分析的结果一致。
为了更加明确所研究体系的π-hole磷键作用的化学键性质,我们对复合物进行了适应性自然密度划分(AdNDP)分析。AdNDP方法是2008年由Zubarev和Boldyrev60提出来的一种分析多中心键的方法,按照整个分子中多中心两电子键表征电子结构,广泛用于分子及簇中的化学键分析。程龙玖等61,62应用AdNDP方法很好地对(Au2S)n及(HgS)n簇中的化学键进行了分析。图S3(Supporting Information)给出了代表性复合物PO2Cl…PCl3(A)、PO2Cl…PH2Cl(A)和PO2Cl…PH2Cl(B)分子间磷键作用的AdNDP轨道图,可以看出PO2Cl…PCl3(A)和PO2Cl…PH2Cl(A)复合物中Pd…Ps间的双中心双电子键(2c-2e)(见图S3(Supporting Information)及PO2Cl…PH2Cl(B)复合物中Cl及O原子的单中心双电子键(1c-2e),即孤对电子。通过对所有体系的AdNDP分析发现,构型A复合物中Pd…Ps间均存在双中心双电子键,电子占据数在1.97以上,而磷原子上不存在孤对电子,说明构型A复合物中Pd…Ps间具有共价键特性。表5列出了构型A复合物中发生分子间相互作用的磷键间2c-2e的电子占据数。对于取代基为NH2的构型B复合物,分子间存在着Pd…N的2c-2e键,其电子占据数为1.99,说明Pd…N间具有共价键特性。对于取代基为卤素的构型B复合物,Pd…X和O…Ps之间不存在2c-2e键,只有卤原子和氧原子的1c-2e键,卤原子在π-hole作用方向的1c-2e键的电子占据数在1.92和1.96之间,氧原子在与PCl3/PH2Cl作用方向的1c-2e键的电子占据数在1.75左右,表5中给出了相应的电子占据数。因此取代基为卤素的构型B复合物分子间具有非共价作用特性。
4 结论
在MP2/aug-cc-pVTZ理论水平上,采用量子化学从头算和电子密度拓扑分析方法,对PO2X和PX3,PH2X(X=F,Cl,Br,CH3,NH2)之间以π-hole作用形成的磷键复合物进行研究,分析了分子静电势、相互作用能、电子密度和能量密度性质、NCI指标、ELF性质等,明确了磷键作用的性质特征。
(1)PO2X中的π-hole与PX3、PH2X中σ-hole的静电势大小决定着分子间相互作用的构型和强度,π-hole作用远大于σ-hole的作用,以PO2X中的π-hole为电子受体形成的磷键复合物有两种稳定构型,分别以Pd…Ps和Pd…X磷键作用为主。
(2)QTAIM、NCI、ELF及AdNDP分析表明,取代基的类型对磷键复合物的性质产生很大影响。当取代基X为给电子基(CH3、NH2)时,磷键具有明显的共价作用特征;当取代基X为吸电子基(F、Cl、Br)时,构型A的磷键表现为共价作用,构型B的磷键表现为非共价或部分共价作用的特征。因此,通过选择取代基可以调控该类磷键作用的性质,这为设计和合成新材料提供了有用的理论信息。
(3)ELF及NBO分析明确了电荷给体和受体轨道间的相互作用,构型B分子间的电荷转移包括PX3/PH2X中X原子上的孤对电子到PO2X中P=O反键空轨道的电荷转移以及PO2X中O原子上的孤对电子到PX3/PH2X中P―X/P―H反键空轨道的电荷转移。电荷给体和受体轨道间相互作用的强度越大,电荷转移量越多,分子间磷键的相互作用越强。
Supporting In fo rm ation:available free of charge via the internetathttp://www.whxb.pku.edu.cn.
Refe ren ces
(1)Politzer,P.;Murray,J.S.ChemPhysChem 2013,14,278.doi: 10.1002/cphc.201200799
(2)Metrangolo,P.;Meyer,F.;Pilati,T.;Resnati,G.;Terraneo,G. Angew Chem.Int.Edit.Engl.2008,47,6114.doi:10.1002/ anie.v47:33
(3)Metrangolo,P.;Neukirch,H.;Pilati,T.;Resnati,G.Accounts Chem.Res.2005,38,386.doi:10.1021/ar0400995
(4)Legon,A.C.Phys.Chem.Chem.Phys.2010,12,7736.doi: 10.1039/c002129f
(5)Scheiner,S.Accounts Chem.Res.2013,46,280.doi:10.1021/ ar3001316
(6)Zahn,S.;Frank,R.;Hey-Hawkins,E.;Kirchner,B.Chemistry 2011,17,6034.doi:10.1002/chem.v17.22
(7)Carré,F.;Chuit,C.;Corriu,R.J.P.;Monforte,P.;Nayyar,N. K.;Reyé,C.J.Organomet.Chem.1995,499,147.doi: 10.1016/0022-328X(95)00318-K
(8)Scheiner,S.Phys.Chem.Chem.Phys.2011,13,13860.doi: 10.1039/c1cp20427k
(9)Solimannejad,M.;Gharabaghi,M.;Scheiner,S.J.Chem. Phys.2011,134,024312.doi:10.1063/1.3523580
(10)Scheiner,S.J.Chem.Phys.2011,134,094315.doi:10.1063/ 1.3562209
(11)Adhikari,U.;Scheiner,S.Chem.Phys.Lett.2012,536,30.doi: 10.1016/j.cplett.2012.03.085
(12)Adhikari,U.;Scheiner,S.J.Phys.Chem.A 2012,116,3487. doi:10.1021/jp301288e
(13)Scheiner,S.;Adhikari,U.J.Phys.Chem.A 2011,115,11101. doi:10.1021/jp2082787
(14)Scheiner,S.Int.J.Quantum Chem.2013,113,1609.doi: 10.1002/qua.v113.11
(15)Del Bene,J.E.;Alkorta,I.;Sanchez-Sanz,G.;Elguero,J. Chem.Phys.Lett.2011,512,184.doi:10.1016/j. cplett.2011.07.043
(16)DelBene,J.E.;A lkorta,I.;Sanchez-Sanz,G.;Elguero,J. J.Phys.Chem.A 2011,115,13724.doi:10.1021/jp2094164
(17)DelBene,J.E.;Alkorta,I.;Sanchez-Sanz,G.;Elguero,J. J.Phys.Chem.A 2012,116,3056.doi:10.1021/jp300763d
(18)A lkorta,I.;Elguero,J.;Del Bene,J.E.J.Phys.Chem.A 2013, 117,4981.doi:10.1021/jp403651h
(19)DelBene,J.E.;Alkorta,I.;Elguero,J.J.Phys.Chem.A 2013, 117,11592.doi:10.1021/jp409016q
(20)DelBene,J.E.;Alkorta,I.;Elguero,J.J.Phys.Chem.A 2013, 117,6893.doi:10.1021/jp4063109
(21)DelBene,J.E.;A lkorta,I.;Elguero,J.Theor.Chem.Acc. 2014,133,1464.doi:10.1007/s00214-014-1464-y
(22)Alkorta,I.;Sanchez-Sanz,G.;Elguero,J.;Del Bene,J.E. J.Phys.Chem.A 2014,118,1527.doi:10.1021/jp411623h
(23)An,X.L.;Li,R.;Li,Q.Z.;Liu,X.F.;Li,W.Z.;Cheng,J.B. J.Mo l.Model.2012,18,4325.doi:10.1007/s00894-012-1445-9
(24)Li,Q.Z.;Li,R.;Liu,X.F.;Li,W.Z.;Cheng,J.B. ChemPhysChem 2012,13,1205.doi:10.1002/cphc.v13.5
(25)Li,Q.Z.;Zhuo,H.;Yang,X.;Cheng,J.B.;Li,W.Z.; Loffredo,R.E.ChemPhysChem 2014,15,500.doi:10.1002/ cphc.v15.3
(26)Del Bene,J.E.;Alkorta,I.;Elguero,J.J.Phys.Chem.A 2014, 118,3386.doi:10.1021/jp502667k
(27)Del Bene,J.E.;Alkorta,I.;Elguero,J.J.Phys.Chem.A 2015, 119,3125.doi:10.1021/acs.jpca.5b00944
(28)Li,Q.Z.;Li,R.;Liu,X.F.;Li,W.Z.;Cheng,J.B.J.Phys. Chem.A 2012,116,2547.doi:10.1021/jp211435b
(29)Alkorta,I.;Elguero,J.;Solimannejad,M.J.Phys.Chem.A 2014,118,947.doi:10.1021/jp412144r
(30)Clark,T.;Hennemann,M.;Murray,J.S.;Politzer,P.J.Mol. Model.2007,13,291.doi:10.1007/s00894-006-0130-2
(31)Murray,J.S.;Riley,K.E.;Politzer,P.;Clark,T.Aust.J.Chem. 2010,63,1598.doi:10.1071/CH10259
(32)Politzer,P.;Murray,J.S.;Clark,T.Phys.Chem.Chem.Phys. 2013,15,11178.doi:10.1039/c3cp00054k
(33)Murray,J.S.;Lane,P.;Clark,T.;Riley,K.E.;Politzer,P. J.Mol.Model.2012,18,541.doi:10.1007/s00894-011-1089-1
(34)Esrafili,M.D.;Mohammadian-Sabet,F.;Solimannejad,M. Struct.Chem.2014,25,1197.doi:10.1007/s11224-014-0392-8
(35)Solimannejad,M.;Ramezani,V.;Trujillo,C.;A lkorta,I.; Sanchez-Sanz,G.;Elguero,J.J.Phys.Chem.A 2012,116,5199.doi:10.1021/jp300540z
(36)Bauza,A.;Ram is,R.;Frontera,A.J.Phys.Chem.A 2014,118, 2827.doi:10.1021/jp502301n
(37)Lang,T.;Li,X.;Meng,L.;Zheng,S.;Zeng,Y.Struct.Chem. 2014,26,213.doi:10.1007/s11224-014-0486-3
(38)Bauza,A.;Mooibroek T.J.;Frontera,A.ChemPhysChem 2015,16,2496.doi:10.1002/cphc.v16.12
(39)Brupbacher-Gatehouse,B.J.Am.Chem.Soc.2000,122,4171. doi:10.1021/ja9938534
(40)Alkorta,I.;Elguero,J.;Del Bene,J.E.J.Phys.Chem.A 2013, 117,10497.doi:10.1021/jp407097e
(41)Boys,S.F.;Bernardi,F.Mol.Phys.1970,19,553.doi: 10.1080/00268977000101561
(42)Frisch,M.;Trucks,G.;Schlegel,H.B.;etal.Gaussian 09, Revision A.02;Gaussian:Wallingford,CT,2009.
(43)Bader,R.F.W.Chem.Rev.1991,91,893.doi:10.1021/ cr00005a013
(44)Matta,C.F.;Boyd,R.J.TheQuantum Theory ofAtoms in Mo lecules;JohnW iley&Sons:New York,2007.
(45)Bone,R.G.A.;Bader,R.F.W.J.Phys.Chem.1996,100, 10892.doi:10.1021/jp953512m
(46)Becke,A.D.;Edgecombe,K.E.J.Chem.Phys.1990,92, 5397.doi:10.1063/1.458517
(47)Silvi,B.;Savin,A.Nature 1994,371,683.doi:10.1038/ 371683a0
(48)Savin,A.;Nesper,R.;Wengert,S.;Fässler,T.F.Angew.Chem. Int.Edit.1997,36,1808.
(49)Li,X.Y.;Huo,S.H.;Zeng,Y.L.;Sun,Z.;Zheng,S.J.;Meng, L.P.Organometallics2013,32,1060.doi:10.1021/om301110j
(50)Bulat,F.A.;Toro-Labbé,A.;Brinck,T.;Murray,J.S.;Politzer, P.J.Mol.Model.2010,16,1679.doi:10.1007/s00894-010-0692-x
(51)Biegler-Kôning,F.J.;Derdau,R.;Bayles,D.;Bader,R.F.W. AIM2000,Version 2.0,2002.
(52)Lu,T.;Chen,F.J.Comput.Chem.2012,33,580.doi:10.1002/ jcc.v33.5
(53)Humphrey,W.;Dalke,A.;Schulten,K.J.Mol.Graph.1996, 14,33.doi:10.1016/0263-7855(96)00018-5
(54)Politzer,P.;Laurence,P.R.;Jayasuriya,K.Environ Health Perspect.1985,61,191.doi:10.1289/ehp.8561191
(55)Politzer,P.;Murray,J.S.Theor.Chem.Acc.2002,108,134. doi:10.1007/s00214-002-0363-9
(56)Koch,U.;Popelier,P.L.A.J.Phys.Chem.1995,99,9747. doi:10.1021/j100024a016
(57)Delanoye,S.N.;Herrebout,W.A.;van der Veken,B.J.J.Am. Chem.Soc.2002,124,11854.doi:10.1021/ja027610e
(58)Johnson,E.R.;Keinan,S.;Mori-Sanchez,P.;Contreras-Garcia,J.;Cohen,A.J.;Yang,W.J.Am.Chem.Soc.2010, 132,6498.doi:10.1021/ja100936w
(59)Contreras-Garcia,J.;Johnson,E.R.;Keinan,S.;Chaudret,R.; Piquemal,J.P.;Beratan,D.N.;Yang,W.J.Chem.Theory Comput.2011,7,625.doi:10.1021/ct100641a
(60)Zubarev,D.Y.;Boldyrev,A.I.Phys.Chem.Chem.Phys.2008, 10,5207.doi:10.1039/b804083d
(61)Cheng,H.;Cheng,L.J.Comput.Theor.Chem.2015,1060,36. doi:10.1016/j.comptc.2015.02.020
(62)Feng,Y.Q.;Cheng,L.J.RSC Adv.2015,5,62543.doi: 10.1039/C5RA06137G
Topological Analyses of Electron Density on π-hole Pnicogen Bonds in PO2X…PX3/PH2X(X=F,Cl,Br,CH3,NH2)Complexes
WANG Yue-Hong LIXiao-Yan ZENG Yan-Li MENG Ling-Peng ZHANG Xue-Ying*
(Key Laboratory of Inorganic Nano-Materials ofHebeiProvince,College ofChemistry and Material Sciences, HebeiNormalUniversity,Shijiazhuang 050024,P.R.China)
Acting as amo lecular linker,the pnicogen bond plays an im portant role in crysta lengineering and supramo lecular synthesis.The structures and properties ofπ-hole pnicogen-bonded com plexes PO2X…PX3and PO2X…PH2X(X=F,Cl,Br,CH3,NH2)we re investigated by ab initio MP2/aug-cc-pVTZ ca lcu lations and topo logica lanalyses ofe lectron density.Two sets o fπ-ho le pnicogen-bonded com plexeswere found on the potentialsurfaces.Type-A complexes have P…P and type-B ones have P…X pnicogen bonds.The atoms-inmolecu les(AIM)theory,electron localization function(ELF)theory,noncovalent interaction(NCI)indexmethod as wellas the adaptive naturaldensity partitioning(AdNDP)app roach were used to expand the nature o f the in teractions considered.The substituent groups strong ly a ffec t the prope rties of pnicogen bond in teractions. Pnicogen bondswere covalent interactions for the e lectron-donating substituents(CH3,NH2),while theywere noncovalent,partly covalentand covalent interactionswhen the substituentswere electron-withdrawing groups. Naturalbond orbita l(NBO)ana lyses indicated that the larger theW iberg bond orderof the pnicogen-bonded interaction,themore cova lent the bond and the greater its strength willbe.In type-B conformations,chargetransfermainly occurs from an X lone pairo f the PX3/PH2Xmolecu le to theπ*(P=O)orbita lof PO2X.
September30,2015;Revised:December23,2015;Published onWeb:December29,2015.
Intermo lecular interaction;Pnicogen bond;π-hole interaction;NCIanalysis; Topologicalanalysis o felectron density;ELF ana lysis
O641
10.3866/PKU.WHXB201512293
*Corresponding author.Email:xueyingzhang@mail.hebtu.edu.cn;Tel:+86-311-80787427.
Theprojectwas supported by the NationalNaturalScience Foundation ofChina(21371045,21372062),Natural Science Foundation of Hebei Province,China(B2014205109),and State Key Program from Ministry of Education of HebeiProvince,China(ZD20131037,ZD20131053).
国家自然科学基金(21371045,21372062),河北省自然科学基金(B2014205109)和河北省教育厅重点项目(ZD20131037,ZD20131053)资助©Editorialofficeof Acta Physico-Chimica Sinica
猜你喜欢
广州化工(2022年19期)2022-11-09 11:30:46
广州化工(2022年18期)2022-10-22 10:27:00
潍坊学院学报(2021年2期)2021-07-22 07:59:12
高等学校化学学报(2021年7期)2021-07-11 16:25:48
国防科技大学学报(2020年6期)2020-12-07 09:25:48
硅酸盐通报(2020年1期)2020-02-25 10:01:30
测绘通报(2019年11期)2019-12-03 01:47:34
赤峰学院学报·自然科学版(2019年5期)2019-09-10 07:22:44
东华大学学报(自然科学版)(2018年1期)2018-06-29 03:34:54
测绘学报(2018年1期)2018-02-27 02:23:07
