
The genetic reference population “Collaborative Cross” is a powerful resource for genetic discoveries and understanding complex genetic traits
2016-09-07 08:30:27GrantMorahan
中国比较医学杂志 2016年7期
Grant Morahan
Centre for Diabetes Research, The Harry Perkins Institute of Medical Research,Centre for Medical Research, University of Western Australia, WA, Australia
The genetic reference population “Collaborative Cross” is a powerful resource for genetic discoveries and understanding complex genetic traits
Grant Morahan
Centre for Diabetes Research, The Harry Perkins Institute of Medical Research,Centre for Medical Research, University of Western Australia, WA, Australia
The Collaborative Cross(CC) is a powerful genetic resource with a wide range of applications in medical research. It is a multiparental genetic reference population, composed of 200 recombinant inbred mouse strains. It was designed to be a resource for rapid mapping of genes that mediate complex traits. The CC has been produced following an intense breeding program that had taken over ten years to complete. It is now becoming available to the research community. While advances in human genetic technologies over the last decade have been substantial, the CC provides the ability to make discoveries not possible with other methods or resources. It can be applied to any subject that can be measured or modelled in the mouse. Here, we discuss a range of applications for which the CC can be used, with examples taken from the early characterization of this powerful resource.
Introduction
Diseases such as cancer, cardiovascular disease, diabetes and dementias pose the greatest public health challenges the world currently faces. These diseases arise from complex interactions between numerous genetic, developmental and environmental factors. Tremendous progress has been made over the last decade in identifying thousands of genetic variants that affect the risk of developing these and other complex genetic diseases (Welter, 2014). However, the mechanisms by which these variants affect disease are mostly unknown, and their detailed examination is difficult, especially in humans. Furthermore, while genome-wide association studies have been successful in finding disease genes, the costs of these studies put them beyond the reach of individual researchers, or for diseases or traits that cannot attract millions of dollars in research support. Gene discovery would be much more rapid and economical if we could circumvent some of the essential tasks that all genetic studies require: 1) recruiting (e.g. patients) or breeding (e.g. F2 animals) a cohort of subjects; 2) characterizing subjects for traits of interest; 3) purifying their DNA samples; 4) typing DNA at informative genetic markers genome-wide; 5) and analyzing the data to find genetic markers that segregate with the trait more often than expected by chance.
Over a decade ago, the groundwork was laid to develop a carefully planned experimental resource that modelled the genetic structure of human populations better than existing resources and that would circumvent the need for repetitive breeding programs, DNA sampling and genotyping (i.e. steps 1, 3, and 4 above). In doing so, it has the potential to trim years of work and hundreds of thousands of dollars from research programs. This resource is a genetic reference population based on a leading model organism, the mouse; it was dubbed The Collaborative Cross (CC) (Churchill 2004).
The concept behind the CC was to exploit the power of Recombinant Inbred (RI) strains, bringing these to state-of-the-art refinement. In the first description of RI strains, Don Bailey wrote that “genetic segregation and recombination provide the very foundation of classical genetics but inbreeding engenders their antithesis though providing great utility… RI strains uniquely exploit these processes concomitantly” (Bailey, 1971). RI strains are produced by crossing two or more inbred strains, and subsequent full sib mating is followed to derive independent parallel strains. Genetic segregation and recombination is achieved in early generations, and recombinant chromosomes are “archived” in later generations as inbreeding proceeds.
Since this insightful paper, RI strains had proven an ideal resource for genetic, immunological and behavioural studies. Providing a virtually unlimited number of genetically identical individuals, they offer key advantages of reproducibility, reduction of genotyping efforts, and wealth of phenotypic characterization. Reproducibility is vital in studying effects if different environmental factors, and conditions with either large variation in responsiveness or with low heritability. RI strains provide the opportunity for reliably testing responses of the same genotype to different environmental conditions. In contrast to most other genetic studies, RI strains need only be genotyped once; these data can be accessed over years, integrating with increased amounts of phenotypic data. In the case of the BXD RI strains, there is now a massive amount of phenotypic and gene expression data available. These data and those for other RI sets are curated and are publicly accessible at the GeneNetworkwebsite (Williams et al., 2001,Chesler et al., 2003).
The rationale for the CC has been discussed extensively (Threadgill 2002, Churchill 2004). The CC was designed to overcome the main limitations of existing RI strain sets: constraints on genetic variability and mapping power. Genetic variability was limited by deriving RI strains from two parental strains; moreover, these parental lines were usually derived from common ancestors, further limiting the genetic repertoire of the derived strains. Mapping power depends on the number of strains available for testing. The resolution of the most widely used sets was typically on the order of 2-4 cM (~4 to 8 Mb); this was not sufficient for efficient candidate gene analyses in the post-genome era. The first problem could be overcome by using eight founder strains, rather than two, and by including wild-derived strains from different continents as founders. The second problem could be overcome by producing larger sets of RI strains. It was envisaged that by increasing the number of CC strains to 1,000, over 135,000 recombination events could be captured,providing a huge increase in mapping resolution, even allowing down-to-the-gene mapping accuracy.
Production of the CC
The CC founder strains were chosen to maximize genetic diversity. Strains whose genomes had just been sequenced (C57BL/6J, A/J, 129S1/Sv1mJ) were selected, as were strains originating from Japan and New Zealand, and which provided models for autoimmunity and obesity (NOD/LtJ and NZO). These were supplemented with wild-derived strains from all three majorMussubspecies: PWK/PhJ aMusmusculusmusculusstrain sourced from Prague; WSB/EiJ, aMusmusculusdomesticusstrain derived from Baltimore; and theMusmusculuscastaneus-derivedCAST/EiJ originating from Thailand. Selection of these founders captured over 90% of the common allelic diversity of the mouse species and across all 1Mb intervals of the genome (Roberts et al. 2007). With the sequencing of the founder strains’ genomes, we now know that on average three non-synonymous polymorphisms are found in over 16,000 genes (Yalcin et al. 2011; Keane et al., 2011). Furthermore, the ECCO database, which integrates the mouse ENCODE data with the CC genomic data has defined over 300,000 variable transcriptional regulatory elements; the eight CC founder strains have more variable sites than the other nine strains that had been sequenced (Nguyen et al. 2013). Thus, natural genetic variation among the strains is a unrivalled source of phenotypic diversity and unlike mutagenesis or knockout programs does not generate debilitating mutations.
An overview of the breeding program is shown in Fig 1. The eight founders were crossed in 56 reciprocal combinations. F1 mice from these crosses were bred at The Jackson Laboratory and used to initiate independent breeding “funnels” at three sites. The breeding scheme was carefully designed to ensure that the strains were derived independently, and that potential genomic interactions between strains were minimized. Each CC strain originated from unique matings of pairs of G2 mice; i.e. no G2 mouse was the ancestor of more than one CC strain.
The three production sites for the CC were in Australia, USA, and Kenya. A total of 916 funnels were established in the Australian site(Morahan, 2008). These strains are currently being moved to Beijing. At ORNL, 650 funnels were initiated; these were later moved to UNC (Chessler, 2008). Supported by the Wellcome Trust, a total of ~120 strains were bred in Kenya, and later moved to Tel Aviv University, Israel (Iraqi, 2008). Although it was anticipated that there would be loss of lines as a result of inbreeding depression, the extent of the loss was unexpected, with less than 200 strains available worldwide. The excessive loss of lines was due in part to lack of funds constraining the number of breeding boxes available for each strain. Nevertheless, a combined total of ~200 CC strains provides a very powerful resource, as discussed below.
The strains were genotyped (Collaborative Cross Consortium, 2012) at 7,500 SNPs using the “Mouse Universal Genotyping Array” (MUGA); the current version of this array provides greater resolution by typing 76,000 SNPs.For each chromosomal segment, founder haplotypes were imputed from the SNP genotypes using tools such as HAPPY (Mott, 2000) or based on normalized intensity values from the genotyping array (Collaborative Cross Consortium, 2012). An example of an imputed genome is shown in Fig 2.
Key findings from studying genotypes of 350 independent CC strains from all three sites were as follows. Founder haplotypes were inherited across strains from all three sites at the expected frequencies, and the decay of linkage disequilibrium was also as expected. Importantly, there was no evidence of gametic disequilibrium in 500 kb windows across the genome. In contrast, common inbred strains showed high and pervasive gametic disequilibrium (i.e. alleles of loci on separate chromosomes could be co-inherited from these strains) that likely resulted from the limiting founder populations from which these strains were derived. Among the inbred strains, no 500-kb window could be guaranteed not to have at least one SNP with very high LD (over 0.75) with unlinked loci in the panel, meaning that association mapping in inbred strains would have high false positive rates. The observed independent inheritance of all genomic intervals vindicated the design of the CC breeding program and indicates that the CC provide an ideal resource for association studies (Collaborative Cross Consortium, 2012).
Capturing extensive genetic diversity results in a wide range of phenotypic variation. The strains are widely variable in the vast majority of all traits examined. This was evident even in the “Pre-CC” i.e. mice characterized during the early phases of the breeding program.
Uses For Rapid Gene Mapping
The CC resource comprises around 200 strains. Will this number be sufficient to meet the aim of not only mapping genes, but doing so with sufficient resolution to identify the causative genetic variants? Simulation studies had been performed using 500 strains, and suggested this could map to within 0.96 cMa single additive locus accounting for 5% of phenotypic variation (Valdar, 2006). It was important to establish whether a smaller number of strains could provide sufficient power for gene mapping. Ram et al. performed a proof-of-principle study, using coat colour data for 120 strains (Ram et al. 2014). Using either a SNP-based approach or an approach based on QTL mapping using founder-derived haplotypes, all four genes mediating six coat colour phenotypes could be mapped. Furthermore, the causative SNP could be identified for three of these genes (the fourth was due to a retroviral insertion). This ability to identify causative variants within minutes of obtaining phenotype data is unprecedented and one of the most powerful features of the CC.
Modifier Genes
Perhaps the most powerful and unique aspect of the CC is its potential to identify genes that protect against diseases. Genes which do not predispose to a disease but which can influence its course or severity are known as “modifier genes”; these are notoriously difficult to identify (Nadeau, J. 2001). However, RI mice are very useful for identification of modifier genes. An outstanding example of this approach comes from a study of modifier genes of breast cancer metastasis. A relevant transgenic mouse was crossed with each of 27 RI lines derived from two parental strains (Lifsted 1998). Despite the smaller size and lower genetic heterogeneity of this panel compared to the CC, this work discovered a gene named “Diasporin” with further work uncovering a set of interacting genes that marked a metastasis-promoting pathway (Crawford 2008). When alleles of humanDiasporaorthologues were investigated in breast cancer patients, differential survival outcomes were successfully found based on patients’ genotypes of theseDiasporagenes (24). These results indicate that modifier genes discovered in the mouse can be directly relevant to human disease.
Ferguson et al. (2013) applied the CC to reveal genes that modified the melanoma-causing effects of two mutant human oncogenes. By careful characterization of various stages of melanoma development, they showed that each step of the pathway was subject to genetic regulation. Some CC strains had median survival times four times higher than the susceptible strains: their naturally occurring genetic variants could prevent the action of multiple copy number mutant transgenes together with the UV mutagen. The CC has the potential for finding protective modifier genes in the many other diseases for which there are established transgenic or otherwise genetically modified mouse models, including a wide range of cancers and neurological diseases. This is another powerful application of the CC, and one that is difficult if not impossible to perform in other systems.
Novel Animal Models
Many researchers would have heard someone declare “the mouse is not a good model” for a particular disease, or that it differs from humans because it does not develop some disease endophenotype. These claims are based on a fallacy: insofar as mice do not display a particular phenotype, this would usually have been ascertained on a small panel of closely related strains. An example of the fallacy is that mice “do not develop type 1 diabetes” but the discovery of the NOD/Lt mouse strain in the 1980s (Makino et al. 1980) has led directly to many insights into the genetics and biology of Type 1 diabetes (reviewed in Driver et al. 2012)reported in over 10,000 publications.
By bringing over 90% of the common genetic repertoire into a single population, and allowing these variants to combine in many different ways, the CC is likely to contain particular combinations of alleles that will predispose to diseases for which there were no previous models. There are already two good examples of novel animal disease models arising from the CC: ulcerative colitis (Rogala et al., 2014) and diabetic retinopathy (Weerasekeraet al. 2015). Other CC strains have been observed to display right ventricular cardiomyopathy, aortic stenosis, achalasia, hypertension and several other conditions (Arnolda, Nguyen, Morahan, unpublished). It should be emphasized that these diseases occur from the interaction of naturally occurring genetic variants, just as common human diseases do, and not from the introduction of crippling genetic modifications. Whenresearchers with expertise in specific disorders characterize more CC strains, doubtless more animal models will be recognized.
New Biological Insights
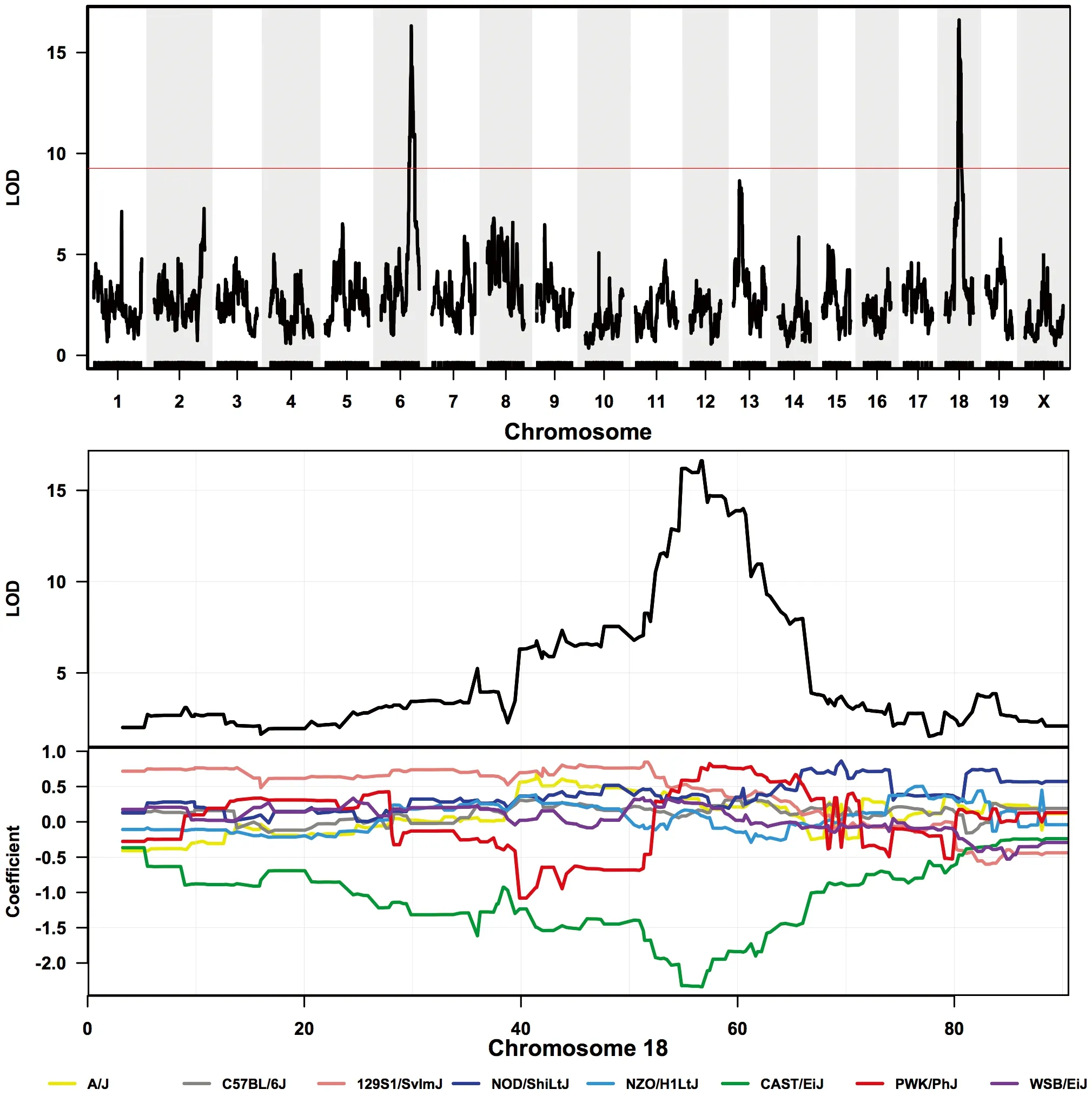
Fig.3 Gene Mapping in The Collaborative Cross.Simulated gene expression data were used to map QTLs. Top panel: genome-wide linkage analysis indicates two significant peaks. Mid panel: LOD Score over chromosome 18. Bottom panel: Contribution of founder strains’ alleles to the chromosome 18 QTL. This analysis shows that the CAST allele differs from all the other founders in mediating the trait of interest. Figures were kindly provided by Dr Ramesh Ram (University of Western Australia).
A unique feature of the RI strains is that each represents a genetically defined individual who is effectively immortal. Unlike outbred populations, phenotypic data can be obtained from any tissue at any developmental stage, under any environmental condition, or induced disease state. The CC can facilitate integration of datafor as wide a range of traits as can be ascertained in the mouse-including but not limited to physiology, immunology, behaviour, immune responses, metabolism, pharmacokinetics, proteomics, developmental variation, disease endophenotypes, epigenetic modifications, microbiome variation, gene expression, etc. Data can be compiled for the same individuals and compared across decades and by researchers around the world. In this respect, the CC facilitates collaborations across time and space. As phenotypic data accumulate in extensive databases, the opportunity arises for observations made by different groups at different times to be correlated and trigger new biological insights.
The largest current dataset is of the BXD RI strain set, and it currently comprises approximately 5,000 phenotypic measurements, including gene expression data for over 30 tissues and cell types. Tools implemented in the WebQTL suite at the GeneNetworkwebsite allow extensive exploration of these data, including mapping genes regulating each of these phenotypes (Williams et al., 2001 and Chesler et al., 2003). For more details, see www.genenetwork.org.
While characterization of the CC is still in its earliest phases, correlation of data has already revealed surprising observations. As an example, bone density measurements of CC strains revealed that, although there was an overall correlation of bone density between male and female mice of the same strain, there were some strains in which the males were osteoporotic and the females osteopetrotic, and vice versa (Figure 3). Obviously, results such as this would not be possible from human studies. Strains which show such strong departure from the expected pattern will provide the basis for further investigations into the molecular basis of the trait of interest, in this case, bone density.
Bioinformatics/Systems Genetics
Systems Genetics (Morahan et al. 2008) analyses integrate gene expression and higher order phenotypic data with underlying genetic variation to define genes and genetic networks that mediate complex traits. The GeneNetwork site has WebQTL tools that allow extensive systems genetics analyses on archived datasets from mouse and other species.
We have integrated the founder strains’ genome sequence variation with data from the ENCODE consortium. The resulting Encode Collaborative Consortium Omnibus (ECCO) databasepermits interrogation of specific chromosome regions, providing custom displays of genetic variation in ENCODE-defined regulatory elements in user-specified sets of strains including the CC founders (Nguyen et al. 2013). This is a useful tool for exploration if a trait has been mapped to a particular locus that contains no nonsynonymous polymorphisms. Complex genetic diseases like type 1 diabetes appear to be mediated by variants that affect transcription of nearby genes rather than changing amino acids in encoded proteins (Ram et al. 2016).
Software tools have been developed for mapping genes and identifying the founder(s) haplotypes that contribute to the trait of interest among CC strains (Ram et al., 2015). Further tools are in development. These include selection of strains with specific founder haplotypes in a region of interest, and integration with the Sanger database of founder genome sequences (www.sysgen.org/GeneMiner). The CC will provide challenges to incorporate new data and inspire further bioinformatic developments.
The Diversity Outcross
A limitation of the CC is the resolution of gene mapping it currently supports: the projected target of 1,000 strains was not reached and the opportunity for recombination events became limiting in the first few generations of inbreeding. This resulted in most strains having large founder haplotypes on most chromosomal regions. A way to increase gene resolution while maintaining the genetic contribution of the same founder strains was to establish a colony of outbred mice derived from CC strains. These “Diversity Outcross” (DO) mice have the advantage over other mouse outcross populations that the only segregating polymorphisms come from the same founders as the CC. This means that the genome sequences of each mouse can be imputed (Svenson, 2012). Compared to the CC strains, they have a high degree of heterozygosity and will harbour shorter founder haplotypes. This feature makes the DO mice useful for high resolution gene mapping applications. They will provide a useful tool to complement studies of the CC. For example, if a QTL is mapped to a region containing many candidate genes, DO mice with recombination events in that region can be selected and tested. This will allow further localization and discrimination of the candidate gene.
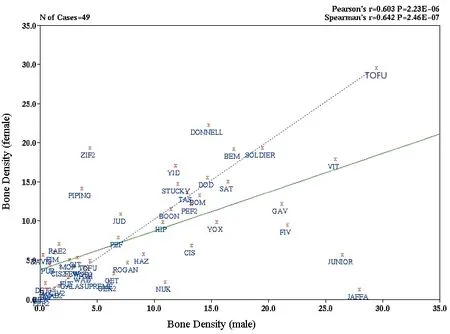
Fig.4 Correlation of Bone Density in Male and Female Mice of CC strains.Femurs from 60 CC strains were characterized by micro-CT and average bone density measurements were calculated for male and female mice for each CC strain from the Australian colony. Data were entered into a modified WebQTL database and traits were correlated. The dotted blue line indicates expected correlation if male and female mice had the same bone density; the green line indicates the actual correlation. Though the overall correlation is significant, it is reduced from the expected due to outlier strains such as JAFFA and ZIF2. These strains have osteopetrotic males but osteoporotic females, and osteopetrotic females but osteoporotic males, respectively. Bone density data were kindly provided by Prof JiakeXu and Jinbo Yuan (University of Western Australia).
The CC as a forerunner of Multi-parental Populations in other species
Recognizing the power of The Collaborative Cross, researchers began production of multiparental populations in other model species. Having the advantage of shorter generation times, these crosses were finished in advance of the CC. Multiparental populations of RI lines have been produced forDrosophila, (King et al., 2012) and many plant species includingArabidopsis, maize, wheat, rice, and chickpea (see review by Huang et al. 2015). Plant versions of the CC are commonlyreferred to asMulti-parent Advanced Generation Intercrosses (MAGIC) and by rapidly identifying genes of agricultural importance, they offer the prospect of improved yields of many crop species.
Concluding Remarks
The CC will be especially valuable to study traits that can only be ascertained by studying the whole animal, in vivo. The 200 extant RI strains provide the opportunity to generate~40,000 genotype combinations each fully defined, reproducible and isogenic within an F1 animal. Such F1 mice will provide even greater opportunities for discovery because unlike the parental CC strains, they will be heterozygous atmost loci, providing the opportunity for investigating effects of heterosis, which is rarely observed at the molecular level (Morahan et al. 2002), and the genetic architecture of particular traits. There is also the prospect of obtaining mice withspecific combinations of genes to create custom genotypes (e.g. heterozygous at loci A, B and C but homozygous for a certain founder strain at loci D and E).
The CC is an extremely powerful and valuable resource. It is not limited to the example applications presented here; its applications are limited by the imagination (and budget!) of researchers. It will provide an enormous amount of data that can be interrogated systematically to enable new insights into mammalian biology that would not be feasible in other systems.
ACKNOWLEDGEMENTS
I am grateful to Prof JiakeXu, Mr Jinbo Yuan and Dr Ramesh Ram for permission to use their bone density data and simulation figures, respectively. The Collaborative Cross strains used for these data were generously provided by Geniad (www.geniad.com).GM’s research is supported by the National Health and Medical Research Council of Australia (Program 1037321 and Project 1069173) and by Diabetes Research Western Australia.
REFERENCES
Bailey DW. Recombinant-inbred strains. An aid to finding identity, linkage, and function of histocompatibility and other genes. Transplantation. 1971 11:325-7.
Chesler EJ1, Wang J, Lu L, Qu Y, Manly KF, Williams RW. Genetic correlates of gene expression in recombinant inbred strains: a relational model system to explore neurobehavioral phenotypes.Neuroinformatics. 2003;1:343-57.
Chesler, E. et al. (2008) The Collaborative Cross at Oak Ridge National Laboratory: developing a powerful resource for systems genetics.MammGenome. 19:382-9.
Churchill G and The Collaborative Cross Consortium (2004) The Collaborative Cross: a community resource for the genetic analysis of complex traits.NatureGenetics,36: 1133-1137.
Collaborative Cross Consortium.The genome architecture of the Collaborative Cross mouse genetic reference population. Genetics. 2012 Feb;190:389-401.
Crawford NP, et al. (2008) The Diasporin Pathway: a tumor progression-related transcriptional network that predicts breast cancer survival.ClinExpMetastasis25:357-69.
Driver JP, Chen YG, Mathews CE.Comparative genetics: synergizing human and NOD mouse studies for identifying genetic causation of type 1 diabetes.
Rev Diabet Stud. 2012 9:169-87
Ferguson B, Ram R, Handoko HY, Mukhopadhyay P, Muller HK, Soyer HP, Morahan G, Walker GJ. Melanoma susceptibility as a complex trait: genetic variation controls all stages of tumor progression. Oncogene. 2015 34:2879-86.
Huang BE, Verbyla KL, Verbyla AP, Raghavan C, Singh VK, Gaur P, Leung H, Varshney RK, Cavanagh CR. MAGIC populations in crops: current status and future prospects. TheorAppl Genet. 2015 128:999-1017.
Iraqi FA, Churchill G, Mott R. (2008) The Collaborative Cross, developing a resource for mammalian systems genetics.MammGenome. 19:379-81.
Keane TM, Goodstadt L, Danecek P, et al. Mouse genomic variation and its effect on phenotypes and gene regulation. Nature. 2011;477:289-294.
King EG, Merkes CM, McNeil CL, Hoofer SR, Sen S, Broman KW, Long AD, Macdonald SJ. Genetic dissection of a model complex trait using the Drosophila Synthetic Population Resource. Genome Res. 2012 Aug;22(8):1558-66.
Lifsted T,et al. (1998) Identification of inbred mouse strains harboring genetic modifiers of mammary tumor age of onset and metastatic progression.IntJCancer. 77:640-4.
Makino S, Kunimoto K, Muraoka Y, Mizushima Y, Katagiri K, Tochino Y. Breeding of a non-obese, diabetic strain of mice.JikkenDobutsu. 1980 29:1-13.
Morahan G, Huang D, Wu M, Holt BJ, White GP, Kendall GE, Sly PD, Holt PG. Association of IL12B promoter polymorphism with severity of atopic and non-atopic asthma in children. Lancet. 2002 360:455-9.
Morahan G, Peeva V, Mehta M and Williams R (2008) Systems genetics can provide new insights into immune regulation and autoimmunity.JAutoimmun. 31: 233-236.
Morahan G, Balmer L and Monley D (2008) Establishment of ‘The Gene Mine’: a resource for rapid identification of complex trait genes.MammGenome. 19: 390-393.
Mott et al. (2000) A new method for fine-mapping quantitative trait loci in outbred animal stocksProcNatlAcadSciUSA, 97:12649-12654.
Nadeau, J. (2001) “Modifier genes in mice and humans”NatureRevGenet2: 165-174
Nguyen C, Baten A, Morahan G. Comparison of sequence variants in transcriptomic control regions across 17 mouse genomes. Database (Oxford). 2014 Mar 18;2014:bau020. doi: 10.1093/database/bau020
Ram R, Mehta M, Balmer L, Gatti DM, Morahan G.Rapid identification of major-effect genes using the collaborative cross.Genetics. 2014 Sep;198(1):75-86.
Roberts A, Pardo-Manuel de Villena F, Wang W, McMillan L, Threadgill DW (2007) The polymorphism architecture of mouse genetic resources elucidated using genome-wide resequencing data: implications for QTL discovery and systems genetics. Mamm Genome 18:473-481
Rogala AR, Morgan AP, Christensen AM, Gooch TJ, Bell TA, Miller DR, Godfrey VL, de Villena FP. (2014) The Collaborative Cross as a resource for modeling human disease: CC011/Unc, a new mouse model for spontaneous colitis.Mamm Genome. 2014 25:95-108.
Svenson KL, Gatti DM, Valdar W, Welsh CE, Cheng R, Chesler EJ, Palmer AA, McMillan L, Churchill GA.High-resolution genetic mapping using the Mouse Diversity outbred population. (2012) Genetics. 190:437-47.
Threadgill DW, Hunter KW, Williams RW (2002) Genetic dissection of complex and quantitative traits: from fantasy to reality via a community effort. Mamm Genome 13:175-178
Weerasekera LY, Balmer LA, Ram R, Morahan G. Characterization of Retinal Vascular and Neural Damage in a Novel Model of Diabetic Retinopathy. Invest Ophthalmol Vis Sci. 2015 Jun;56(6):3721-30.
Valdar W., Flint J., Mott R., 2006. Simulating the collaborative cross: power of quantitative trait loci detection and mapping resolution in large sets of recombinant inbred strains of mice. Genetics 172: 1783-1797
Welter D, MacArthur J, Morales J, et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations.NucleicAcidsRes. 2014; 42 (Database issue). D1001-6. doi, 10.1093/nar/gkt1229.
Williams RW1, Gu J, Qi S, Lu L.The genetic structure of recombinant inbred mice: high-resolution consensus maps for complex trait analysis. Genome Biol. 2001;2(11):RESEARCH0046. Epub 2001 Oct 22.
Yalcin B., Flint J., Mott R., 2005. Using progenitor strain information to identify quantitative trait nucleotides in outbred mice. Genetics 171: 673-681
R-332【文献标识码】 A
1671-7856(2016)07-0001-10
10.3969.j.issn.1671-7856.2016.07.001
2016-05-31
[Corresponding Author]Prof Grant Morahan, Centre for Diabetes Research, Level 7, QQ Block Sir Charles Gairdner Hospital Nedlands, 6009 WAAustralia Tel: +61-8-6151 0756,E-mail: grant.morahan@uwa.edu.au
编者按:在常用实验动物中,小鼠资源最为丰富,具有各种不同特点的近交品系、突变品系、封闭群以及基因工程小鼠等,然而,随着对疾病的深入研究,发现采用单一遗传背景的小鼠无法有效复制人类多基因、多因素的相互作用引起的复杂性疾病,如高血压、糖尿病、心脏病、肿瘤等。实验动物育种专家培育了一种能够很好的模拟人群多基因、多因素作用的复杂性状特点的小鼠资源,复杂性状遗传CC(Collaborative Cross, CC)小鼠。CC小鼠由八种纯系的实验室常用小鼠品系重组近交而来,这八种品系小鼠的多样性等位基因可覆盖小鼠全基因组90%的区域,品系间总共存在36M的单核苷酸多态性(SNPs)。该小鼠资源能体现不同小鼠亚种的遗传学变异,具有高度遗传多样性、大种群规模等可以模拟人群的重要特征。每种小鼠品系可以通过软件进行基因组溯源,经过系统遗传学的基因连锁分析,有望鉴定与人群复杂性状疾病相关的遗传学基础,潜在靶点分析,为临床精准医疗提供强有力的工具。
本刊将陆续推出系列文章,介绍CC小鼠,和它的应用情况。敬请读者关注。
猜你喜欢
贵州畜牧兽医(2023年3期)2023-06-29 07:07:28
农技服务(2023年2期)2023-03-15 00:43:08
今日农业(2021年11期)2021-08-13 08:53:24
园林科技(2020年2期)2020-01-18 03:28:18
福建基础教育研究(2019年10期)2019-05-28 08:27:04
求学·理科版(2017年3期)2017-04-27 22:06:36
中国中医药现代远程教育(2014年23期)2014-03-01 04:33:43
作物研究(2014年6期)2014-03-01 03:39:03
遗传(2014年3期)2014-02-28 20:59:30
遗传(2014年3期)2014-02-28 20:58:49
