
基于CO I与ITS序列的杂鳞库蚊复组(双翅目:蚊科)分子系统发育
2016-08-23 03:13:46赵文静张春林陈汉彬
环境昆虫学报 2016年4期
赵文静,张春林,陈汉彬,张 晶,刘 彬
(贵州医科大学生物学教研室,贵阳 550004)

基于CO I与ITS序列的杂鳞库蚊复组(双翅目:蚊科)分子系统发育
赵文静,张春林*,陈汉彬,张晶,刘彬
(贵州医科大学生物学教研室,贵阳 550004)
使用杂鳞库蚊复组CO I部分序列和ITS序列构建分子发育树,并基于CO I序列计算该复组种内和种间的Kimura-two-Parameter (K2P)距离,探讨环带库蚊的分类地位和杂鳞库蚊复组内各亲缘种的系统发育关系。环带库蚊和杂鳞库蚊的种间K2P距离为0.24%-0.72%,支持“环带库蚊是杂鳞库蚊的同物异名”这一观点;杂鳞库蚊(环带库蚊)和伪杂鳞库蚊、三带喙库蚊的种间K2P距离为4.41%-9.68%,同时分子系统树显示各个种分别聚类,互为姐妹群,再次证明三者互为独立的种;环带库蚊和杂鳞库蚊聚类的分支最接近树的端部,三带喙库蚊分支最接近树的基部,提示三带喙库蚊最早发生分化,而杂鳞库蚊(环带库蚊)最晚发生分化;采集自日本的三带喙库蚊种内K2P距离为0.48%-2.68%,而它们与采集自中国、印度的该种K2P距离为4.17%-6.76%,日本产三带喙库蚊聚集成一支,并与中印产地的聚类分支互为姐妹群,这些结果提示日本的三带喙库蚊有种下,甚至种级分化的趋势。
杂鳞库蚊复组;核酸序列;K2P距离;分子系统发育
杂鳞库蚊复组Culexvishnuicomplex隶属于库蚊属海滨库蚊组Culexsitiensgroup,主要分布于东洋界,并向古北界延伸。目前记载有4个种:环带库蚊C.annulusTheobald, 1901,杂鳞库蚊C.vishnuiTheobald, 1901,伪杂鳞库蚊C.pseudovishnuiColless, 1957和三带喙库蚊C.tritaeniorhynchusGiles, 1901。其共同特征是:阳茎侧板内叶(复支)密生小刺;中叶分4-5个指环突;外叶的背、腹齿约等大;载肛突基内侧有侧楔;喙和跗有淡色环。其中三带喙库蚊已经被多次证实是日本脑炎病毒(Japaneseencephalitisvirus)的主要传病媒介。该复组与医学关系密切,因此自报道以来,便引起国内外学者的关注。但是由于杂鳞库蚊复组的各亲缘种在形态上较相近,各个种的变异幅度较大,所以它的分类历史上一直存在着不同的观点和鉴定困难。譬如环带库蚊在头顶正中的竖鳞颜色、翅长、幼虫头毛1-C等均有不同程度的变异,因此在对其变异规律未加研究的情况下,很可能把种内居群或个体误定为新种——谢麟阁和廖定西(1956)报告的混同库蚊CulexpermixtusHsieh & Liao, 1956就属于这种情况。我国杂鳞库蚊复组曾报告过7个种和1个亚种,分类比较混乱,后经陈汉彬(1980)进行系统整理和澄清,认为杂鳞库蚊复组在我国境内只有3种,即环带库蚊、伪杂鳞库蚊和三带喙库蚊。不过遇到形态变异较大的情况,仍然不易于分辨。另外,对于“环带库蚊是杂鳞库蚊的同物异名”这一说法,一直存在着争议。自Theobald(1901)对环带库蚊描述后, Colless(1957)、Bram(1967)、Reuben(1969)、Miyagi和Iha(1970)、Sirivanakarn(1975,1976) 、陈汉彬(1980)先后从形态学上对其再描述,但没有达成统一意见。例如,Reuben(1969)根据印度马德拉斯(杂鳞库蚊的模式标本产地)标本,认为杂鳞库蚊和环带库蚊的幼虫没有差别,建议将环带库蚊作为杂鳞库蚊的同物异名处理。随后Sirivanakarn (1975)也基于同样理由建议把环带库蚊作为杂鳞库蚊的亚种或地理型。然而陈汉彬(1980)认为,幼虫的同一性并不能排除成虫的差异性,杂鳞库蚊成虫喙、翅和腿上具浓密的淡鳞麻点是它与环带库蚊的重要鉴别特征,因此建议将“环带库蚊是杂鳞库蚊的同物异名”这一说法做存疑处理,在我国继续沿用环带库蚊这一名称。
自核酸序列被广泛应用于蚊科的系统发育研究以来,其呈现的分子多态性和构建的分子树,成为分类学研究的有力依据,同时也为鉴别杂鳞库蚊复组的近缘种提供了便利。例如Dhananjeyan等 (2010)提取并扩增印度地区杂鳞库蚊、伪杂鳞库蚊和三带喙库蚊的虫卵、幼虫和成虫的ITS 2序列,根据它们的ITS 2凝胶电泳条带位置差异,实现简单、快速的种间鉴别。Kumar等(2007)测定印度地区蚊类22个种的mtDNA CO I序列,构建NJ树,这种方法成功鉴别出绝大部分蚊类,包括杂鳞库蚊、伪杂鳞库蚊和三带喙库蚊。Toma等(2000)利用PCR法扩增杂鳞库蚊、伪杂鳞库蚊、三带喙库蚊等蚊类的ITS 1-5.8S-ITS 2序列,有效的鉴别出杂鳞库蚊的这3个近缘种。Miller等(1996)测定14种库蚊(其中包含三带喙库蚊和伪杂鳞库蚊)的ITS序列,并基于此构建分子树,能够区分出三带喙库蚊和伪杂鳞库蚊。遗憾的是,这些文献都缺乏环带库蚊的样本,不能够解决“环带库蚊是否为杂鳞库蚊的同物异名”这一关键问题。反之,涉及到环带库蚊的分子系统发育研究又缺乏杂鳞库蚊的样本。例如杨明等(2003)利用RAPD-PCR技术获得环带库蚊、伪杂鳞库蚊和三带喙库蚊ITS 2序列,根据它们的长度和数量成功鉴别出三者。
随着生物信息公共数据库的推广和应用,利用互联网整合现有资源并进行二次分析的做法,既可以克服人力物力缺乏带来的标本采集困难,又可以避免重复性实验,能够利用更广泛的数据获得更全面的结论。鉴于此,本研究从GenBank数据库中收集大部分杂鳞库蚊复组mtDNA CO I和ITS序列,并基于这些序列进行分子系统发育研究,讨论杂鳞库蚊复组内各亲缘种的分类地位和系统发育关系。另外,由于公共数据库缺乏环带库蚊相关序列,所以进行补充测定。
1 材料与方法
1.1环带库蚊mtCO I序列,rDNA ITS序列的获取
GenBank数据库缺乏环带库蚊CO I序列和ITS 1 序列,所以根据现有杂鳞库蚊复组序列的长度和覆盖区域,进行补充测定。数据库中的CO I序列均为部分序列,本文设计扩增的环带库蚊CO I片段完全覆盖并远远大于数据库中被使用的最短序列。另外由于含ITS 1的序列均以“18S部分序-ITS 1-5.8S-ITS 2-28S部分序列”的形式存在,为获得同一个样本的ITS序列(包含ITS 1和ITS 2序列),进行同样区域的扩增。
环带库蚊于2010年8-9月和2013年7月,自贵阳三江农场稻田内采集到幼虫,经羽化、鉴定后,置无水乙醇,-20℃保存。采用常规酚-氯仿法提取环带库蚊4龄幼虫线粒体DNA,并调整浓度为50 ng/mL。
PCR扩增mtDNA CO I部分基因,沿用Kumar等(2007)设计的引物。引物序列为CO I-For:5′-GGATTTGGAAATTGATTAG TTCCTT-3′;CO I-Rev:5′-AAAAATTTTAATTCCAGTTGGAACAGC-3′。PCR反应体系为:1 × PCR Buffer, 1.25 mM Mg2 +, 0.4 mM dNTPs, 0.08 pM引物, 4 ng/μL总DNA 模板, 0.02 U /μL TaKaRa Taq酶。扩增条件依次为: 95℃预变性5 min;94℃变性40 s, 51℃退火1 min, 72℃延伸1 min;循环35次后,72℃延伸10 min;4℃保温。
PCR扩增rDNA ITS序列,结合软件Primer premier 6.0和Oligo 7.27设计引物。引物序列为:ITS-For:5′-ATTTGAATCG CTGAAGTTG-3′;ITS-Rev:5′-GTAGTCACACATTATTTGAGGC-3′,由上海生物工程技术服务有限公司合成。PCR反应体系为:1 × PCR Buffer, 1.25 mM Mg2+, 0.4 mM dNTPs, 0.08 pM引物, 4 ng/μL总DNA 模板, 0.02 U/μL TaKaRa Taq酶。扩增条件依次为: 95℃预变性5 min;94℃变性40 s, 54℃退火1 min,(此后9个循环每个循环递减0.5℃,至第11个循环维持48℃的退火温度),72℃延伸1 min;共循环35次后,72℃再延伸7 min;4℃保温。
所有PCR产物都经1%琼脂糖凝胶电泳、纯化、回收后送至上海生物工程技术服务有限公司测序。测序结果经NCBI Blastn检验,确定为杂鳞库蚊复组的基因。它们的登录号见表1。
1.2序列信息
序列信息详见见表1。
1.3序列处理
为确保序列的有效性,作者逐条校对、查阅了表1中所有序列的登录信息和支撑文献,并做BLASTn比对; 其次,在Barcode of Life Data System 网站(http://www.boldsystems.org/)进行验证。最后选用BioEdit软件编辑序列。
对于本次所获得环带库蚊CO I部分序列,首先根据测序峰值进行人工校对,切除序列两端不可信区域。然后使用Clustal W对所有CO I序列进多重比对,切齐序列两端;以高保守区5′-GCAATAAATAATATAAGTTTTTGAATA-3′为起始端,切齐5′端,根据所用的最短序列切齐3′端。对于ITS序列,除去编码rDNA 18S、5.8S和28S的序列,再拼接ITS 1和ITS 2序列,得到同一个样本的ITS全序列。
1.4系统发育分析
选用MAGE 6.0分析DNA序列的组成及变异情况,并用Kimura-two-Parameter (K2P) 模型估计分化距离(K2P距离);使用Model test 3.7估算构建ML树的最优模型,再以此模型建ML树;使用Jukes-Cantor模型对平均K2P距离进行检测,若平均距离P<1,则表示适合构建NJ建树;使用PAUP 4.0构建ML树。
2 结果与分析
2.1环带库蚊CO I部分序列 和ITS全序列
环带库蚊4个个体CO I部分基因,GenBank登陆号为KC876676-KC876679,长度为649-672 bp,GC含量为31.6%-32.0%,G含量为16.2%-16.3%,表现出很强的反G偏倚,符合昆虫mtDNA的特点。种内4个个体仅1个个体第530位碱基较其他序列发生转换A→G。环带库蚊2个个体SJ-N1和SJ-N2的“18S部分序列ITS 1-5.8S-ITS 2-28S部分序列”序列,GenBank登陆号分别为KF499144,KF4991445,长度均为903 bp,其中ITS 1为380 bp,ITS 2为253 bp,它们与早期登录的环带库蚊rDNA序列AF453488所推测的ITS 1和ITS 2相比,SJ-N1样本在ITS 2序列的第190, 191位GC碱基上发生缺失;SJ-N2样本在ITS 1序列的250位碱基上发生转换T→C, 在ITS 2序列的第248、249位AC碱基上发生缺失。环带库蚊CO I和ITS序列的碱基组成见表2。

表1 杂鳞库蚊复组序列信息
续上表
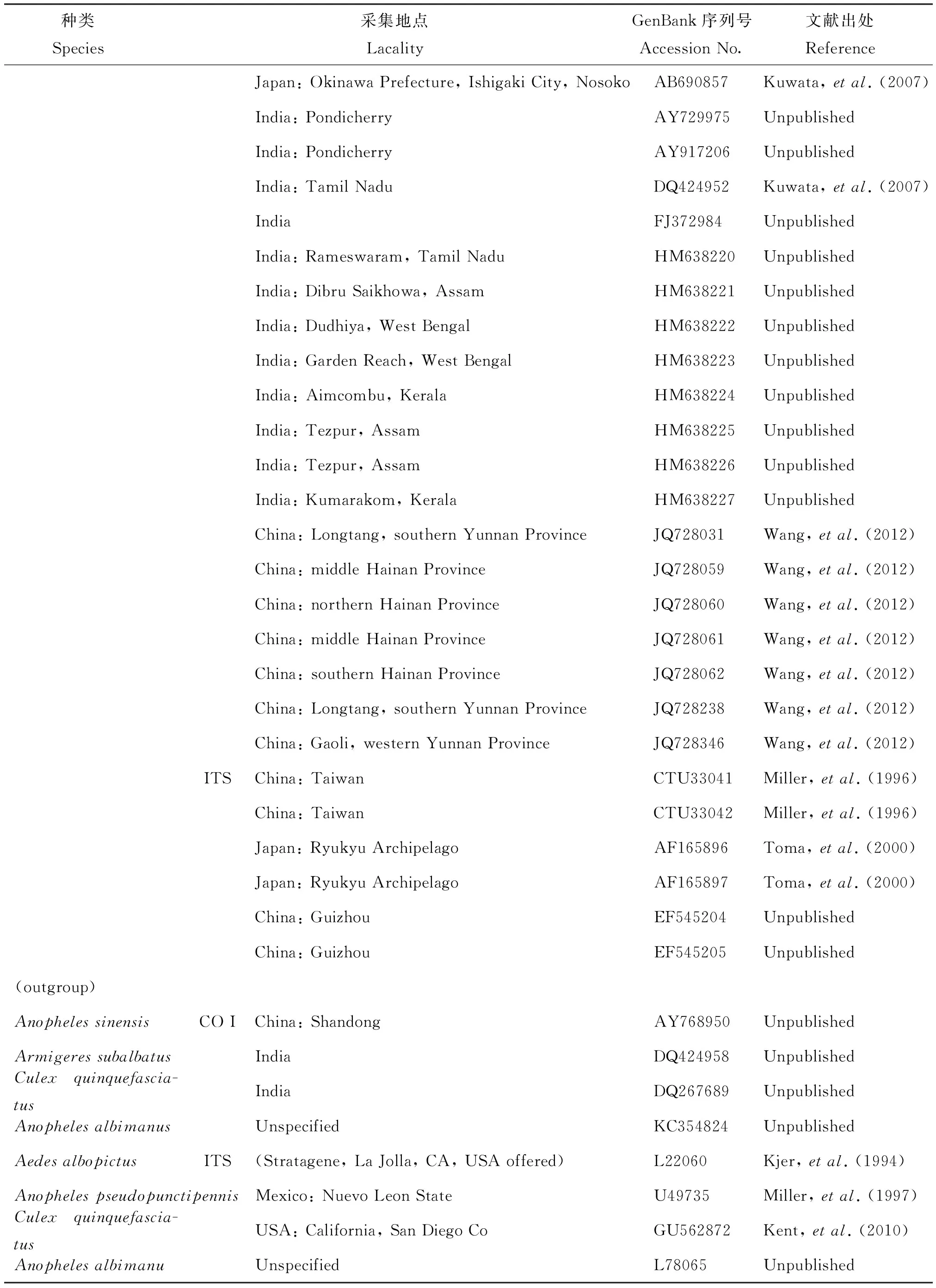
种类Species采集地点LacalityGenBank序列号AccessionNo.文献出处ReferenceJapan:OkinawaPrefecture,IshigakiCity,NosokoAB690857Kuwata,etal.(2007)India:PondicherryAY729975UnpublishedIndia:PondicherryAY917206UnpublishedIndia:TamilNaduDQ424952Kuwata,etal.(2007)IndiaFJ372984UnpublishedIndia:Rameswaram,TamilNaduHM638220UnpublishedIndia:DibruSaikhowa,AssamHM638221UnpublishedIndia:Dudhiya,WestBengalHM638222UnpublishedIndia:GardenReach,WestBengalHM638223UnpublishedIndia:Aimcombu,KeralaHM638224UnpublishedIndia:Tezpur,AssamHM638225UnpublishedIndia:Tezpur,AssamHM638226UnpublishedIndia:Kumarakom,KeralaHM638227UnpublishedChina:Longtang,southernYunnanProvinceJQ728031Wang,etal.(2012)China:middleHainanProvinceJQ728059Wang,etal.(2012)China:northernHainanProvinceJQ728060Wang,etal.(2012)China:middleHainanProvinceJQ728061Wang,etal.(2012)China:southernHainanProvinceJQ728062Wang,etal.(2012)China:Longtang,southernYunnanProvinceJQ728238Wang,etal.(2012)China:Gaoli,westernYunnanProvinceJQ728346Wang,etal.(2012)ITSChina:TaiwanCTU33041Miller,etal.(1996)China:TaiwanCTU33042Miller,etal.(1996)Japan:RyukyuArchipelagoAF165896Toma,etal.(2000)Japan:RyukyuArchipelagoAF165897Toma,etal.(2000)China:GuizhouEF545204UnpublishedChina:GuizhouEF545205Unpublished(outgroup)AnophelessinensisCOIChina:ShandongAY768950UnpublishedArmigeressubalbatusIndiaDQ424958UnpublishedCulexquinquefascia-tusIndiaDQ267689UnpublishedAnophelesalbimanusUnspecifiedKC354824UnpublishedAedesalbopictusITS(Stratagene,LaJolla,CA,USAoffered)L22060Kjer,etal.(1994)AnophelespseudopunctipennisMexico:NuevoLeonStateU49735Miller,etal.(1997)Culexquinquefascia-tusUSA:California,SanDiegoCoGU562872Kent,etal.(2010)AnophelesalbimanuUnspecifiedL78065Unpublished
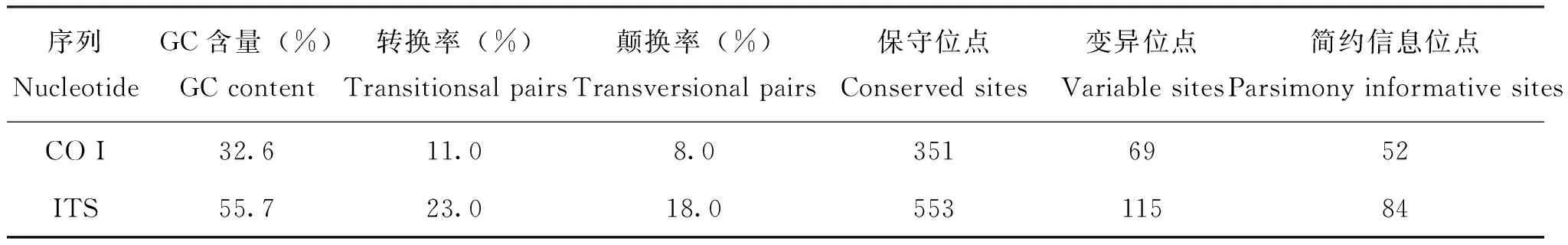
表2 环带库蚊CO I和ITS序列的碱基组成
2.2分子系统发育树
使用Jukes-Cantor模型分别估算CO I和ITS序列的平均K2P距离,P值结果分别为0.052和0.354,P<1,表示三组核酸序列数据均适合构建NJ树。使用Model test 3.7估算构建ML树的最优模型,基于CO I和ITS序列的最优建树模型均为GTR+I+G,所以以此模型构建ML树。Bootstrap=1000。结果如图1、图2所示。
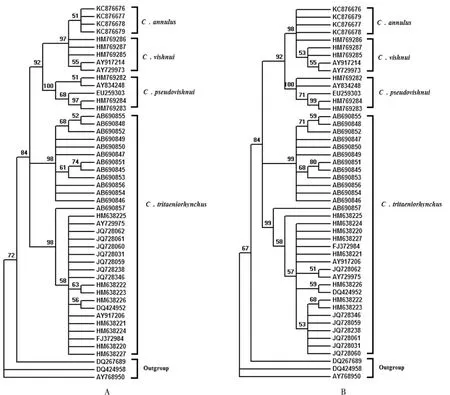
图1 基于CO I序列构建的杂鳞库蚊复组系统发育树,图A最大似然法(ML)、图B邻接法(NJ),置信度为50%Fig.1 Maximum-likelihood (phylogenies A) and Neighbor-Joining(phylogenies B) trees for the Culex vishnui complex are based on CO I gene, with cut-off value of 50%
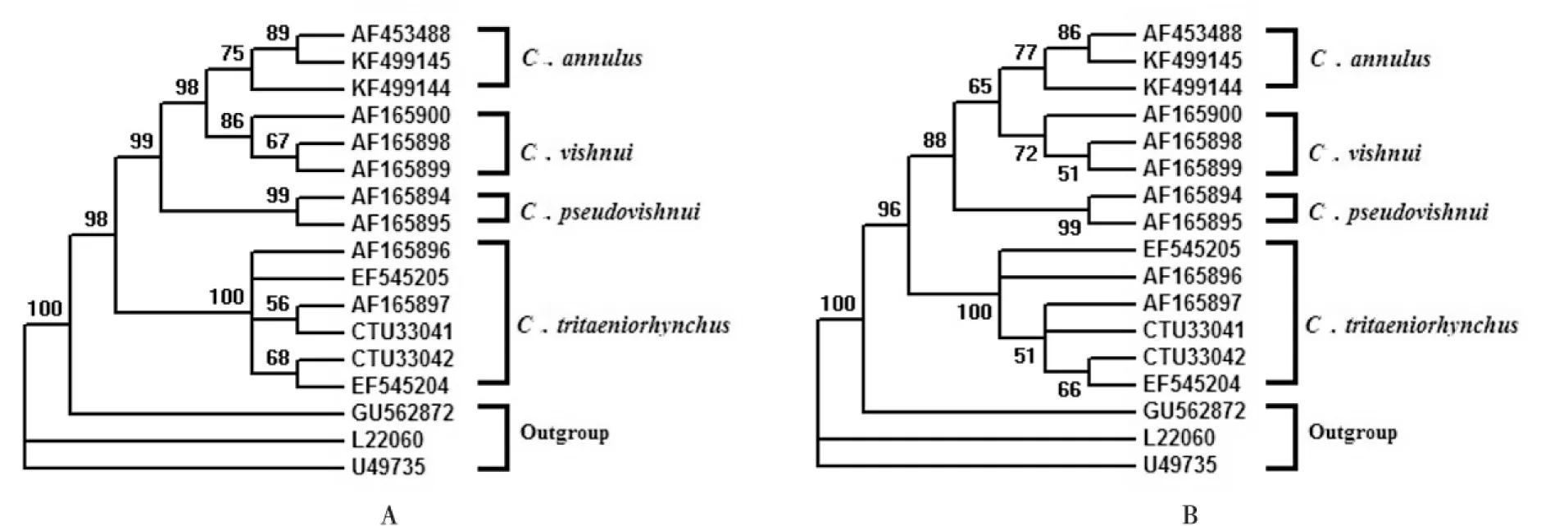
图2 基于ITS序列构建的杂鳞库蚊复组系统发育树,图A最大似然法(ML)、图B邻接法(NJ),置信度为50%Fig.2 Maximum-likelihood (phylogenies A)and Neighbor-Joining(phylogenies B) trees for the Culex vishnui complex are based on ITS sequence, with cut-off value of 50%
2.3基于CO I序列的杂鳞库蚊复组的K2P距离
使用CO I序列计算杂鳞库蚊复组的K2P距离并生成表格,随后以种为单元统计数据,结果如表3所示。
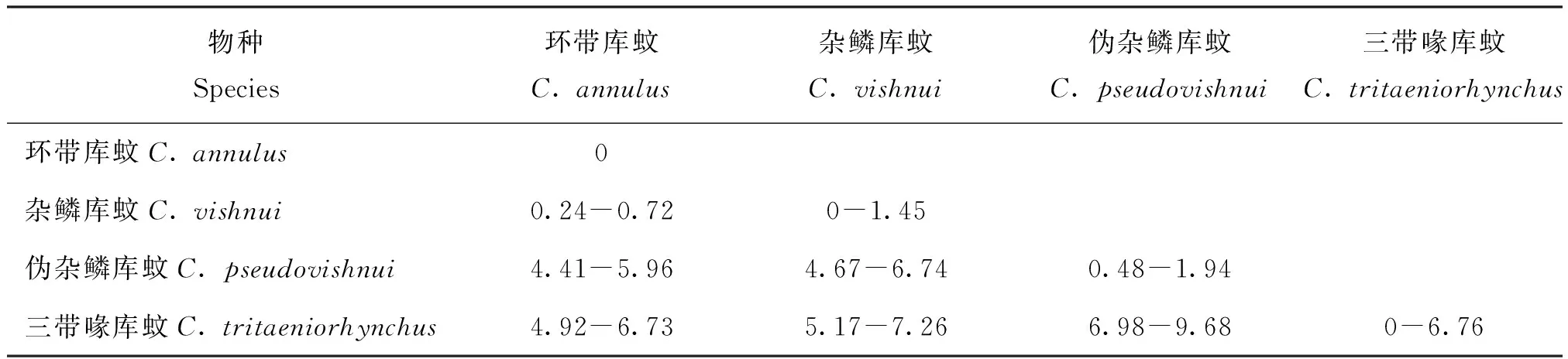
表3 基于CO I序列的杂鳞库蚊复组的K2P距离(%)
2.4基于CO I序列的三带喙库蚊的K2P距离
三带喙库蚊的CO I序列采集自日本、中国和印度各地区,计算它们之间的K2P距离,结果如表4。
3 结论与讨论
3.1关于分子发生树
使用CO I和ITS序列构建的ML树和NJ树(见图1、图2),具有基本一致的拓扑结构。总的来看,杂鳞库蚊复组构成一单系群,各个种分别聚集,互为独立的种,这与形态学分类的结果一致。其中环带库蚊和杂鳞库蚊总能聚成一支,该分支与伪杂鳞库蚊分支互为姐妹群,前三者形成的分支与三带喙库蚊分支互为姐妹群。可见环带库蚊和杂鳞库蚊享有更近的节点,这说明它们享有更近的假想祖先,拥有更近的亲缘关系。同理,它们与伪杂鳞库蚊的亲缘关系较近,而与三带喙库蚊较远。
其次,环带库蚊与杂鳞库蚊聚类的分支更接近树的端部,说明它们发生分化的时间较晚;三带喙库蚊分支最接近树的基部,说明三带喙库蚊是杂鳞库蚊复组内最早发生分化的种。
再次,CO I分子系统树显示三带喙库蚊分为两个大支,一支由登录号为AB690845-AB690855的样本构成,采集于日本各地区,另一支采集于中国和印度。这两大支与杂鳞库蚊复组的其他三个种的分支互为姐妹群,提示日本的三带喙库蚊有种下,甚至种级分化的趋势。
3.2关于种间K2P距离
Hebert等(2002),通过统计7属共200种鳞翅目昆虫CO I 序列的K2P距离,提出在分子水平上界定鳞翅目的种间K2P距离是>3%。这一结果在他们的实践中得到100%的成功,因此他们认为这种方法可以推广用于其他物种的种的鉴定。Wang等(2012)出于同样的思路,使用CO I序列对中国常见蚊类进行鉴定。他们统计了15属122种蚊类CO I序列的K2P距离,并绘制分子发生树(分为按蚊属、库蚊属和其他蚊类三棵树)。结果表明,超过98%的蚊类遵循种间K2P距离>2%,而种内K2P距离<2%的规律,其中包括隶属于杂鳞库蚊复组的三带喙库蚊。综上我们推测,CO I序列的种间K2P距离2%,可以作为杂鳞库蚊复组在分子水平上的定种阈值。
如表3所示,杂鳞库蚊和环带库蚊的K2P距离是0.24%-0.72%,远小于2%(低了一个数量级),结果支持“环带库蚊是杂鳞库蚊的同物异名”这一观点。由于杂鳞库蚊与环带库蚊报告于同一期期刊,但因为报告杂鳞库蚊的篇幅(355页)略前于环带库蚊的(358页)(Reuben, 1969),所以依照国际命名法规,将环带库蚊改称为杂鳞库蚊是可取的。
此外,就种间K2P距离来说,杂鳞库蚊(含环带库蚊)与伪杂鳞库蚊、三带喙库蚊的K2P距离分别为4.41%-6.74%、4.92%-7.26%;伪杂鳞库蚊和三带喙库蚊的K2P距离为6.98%-9.68%,结果均大于2%两倍以上,再次证明杂鳞库蚊复组3个亲缘种互为独立的种。
就种内K2P距离而言,杂鳞库蚊样本采集于印度大陆多处,环带库蚊样本采自于中国贵阳,它们的种内K2P距离分别为0.53%和0;伪杂鳞库蚊采自于印度大陆和日本各岛,它的种内K2P距离为1.36%。上述两个种的种内K2P距离都未超过2%。
例外的是三带喙库蚊(如表4所示),它们采集于中国、印度和日本,种内K2P距离在0-6.76%,整体来看并不遵守“2%规律”,但实际上主要是来自于日本的三带喙库蚊打破了这一规律。按照CO I分子树的结构,将三带喙库蚊划分为日本产地和中、印产地两个类群,两个类群内部的K2P距离分别为0.48%-2.68%,和 0-2.68%,基本遵循“2%规律”,属于种内的变化范围。但是日本产地的类群,与中、印产地的K2P距离,即两个类群之间的K2P距离为4.17%-6.76%。这一数值近似于杂鳞库蚊(含环带库蚊)与伪杂鳞库蚊之间的K2P距离(4.41%-6.74%),即种间的分化距离。综上,我们推测日本的三带喙库蚊有种下,甚至种级分化的趋势。形成这一结果的可能原因是,日本列岛地处欧亚大陆板块、北美洲板块、太平洋板块及菲律宾板块4个板块的交界处,地形地貌比较复杂,此外它与中国、印度所处的欧亚大陆有海洋间隔,为适应环境,三带喙库蚊在扩散的途中产生较大的变异,甚至发生种下或种级的分化。不过更确凿的结论还需要进一步调查和实验研究才能获得。
References)
Bram RA.Contributions to the mosquito fauna of southeast Asia-II.The genusCulexin Thailand (Diptera: Culicidae)[J].ContributionsoftheAmericanEntomologicalInstitute(Ann Arber), 1967, 2(1): 1-296.
Colless DH.Note on the culicine mosquitoes of Singapore[J].AnnalsofTropicalMedicine&Parasitology, 1957, 51: 87-101.
Chen HB.Study on theCulexvishnuisubgroup in China with discussions on the taxonomic status ofC.neovishnuiLien,C.permixtusHsieh & Liao andC.cheniHo.[J].ActaEntomologicaSinica, 1980, 23(4): 434-440. [陈汉彬. 我国杂鳞库蚊亚组的初步研究[J].昆虫学报, 1980, 23(4): 434-440]
Dhananjeyan KJ, Paramasivan R, Tewari SC,etal.Molecular identification of mosquito vectors using genomic DNA isolated from eggshells, larval and pupal exuvium[J].Trop.Biomed., 2010, 27 (1): 47-53.
Hebert PDN, Cywinska A, Ball SL,etal. Biological identifications through DNA barcodes[J].TheRoyalSociety, 2002, 270: 313-321.
Kent RJ,Deus S,Williams M,etal.Development of a multiplexed polymerase chain reaction-restriction fragment length polymorphism (PCR-RFLP) assay to identify common members of the subgeneraCulex(Culex) andCulex(Phenacomyia) in Guatemala[J].Am.J.Trop.Med.Hyg., 2010, 83(2): 285-291.
Kjer KM, Baldridge GD, Fallon AM.Mosquito large subunit ribosomal RNA: Simultaneous alignment of primary and secondary structure[J].Biochim.Biophys.Acta, 1994, 1217(2): 147-155.
Kumar NP, Rajavel AR, Natarajan R,etal.DNA barcodes can distinguish species of indian mosquitoes (Diptera: Culicidae)[J].J.Med.Entomol., 2007, 44(1): 1-7.
Kuwata R,Hoshino K,Isawa H,etal.Establishment and characterization of a cell line from the mosquitoCulextritaeniorhynchus(Diptera: Culicidae)[J].InVitroCell.Dev.Biol.Anim., 2012, 48(6): 369-376.
Miller BR, Crabtree MB, Savage HM.Phylogeny of fourteenCulexmosquito species, including theCulexpipienscomplex, inferred from the internal transcribed spacers of ribosomal DNA[J].InsectMol.Biol.,1996, 5(2): 93-107.
Miller BR, Crabtree MB, Savage HM.Phylogenetic relationships of the Culicomorpha inferred from 18S and 5.8S ribosomal DNA sequences. (Diptera:Nematocera)[J].InsectMol.Biol., 1997, 6 (2): 105-114.
Miyagi I and Iha S.Notes onCulex(Culex)neovishnuiLien,1968 from the Ryukyus and Japan proper(Diptera: Culicidea)[J].Trop.Med. (Nagasaki), 1970,12:71-78.
Reuben R.A re-description ofCulexvishnuitheo with notes onC.psedovishnuiColless andC.tritaeniorhynchusCiles from southern India[J].Bull.Entomol.Res.,1969, 58:643-652.
Sirivanakarn S.The systematics ofCulexvishnuicomplex in southeast Asia with the diagnosis of three common species (Diptera: Culicidae)[J].MosquitoSystematics,1975, 7(1): 69-85.
Sirivanakarn S.Medical Entomology Studies-Ⅲ. A Revision of the SubgenusCulexin the Oriental Region (Diptera: Culicidae)[C].ContributionsoftheAmericanEntomologicalInstitute, 1976, 12(2): 1-186.
Toma T,Miyagi I,Crabtree MB,etal.Identification ofCulexvishnuisubgroup (Diptera: Culicidae) mosquitoes from the Ryukyu Archipelago, Japan: Development of a species-diagnostic polymerase chain reaction assay based on sequence variation in ribosomal DNA spacers[J].J.Med.Entomol., 2000, 37 (4): 554-558.
Wang G, Li C, Guo X,etal. Identifying the main mosquito species in china based on DNA barcoding[J].PLoSONE, 2012,7(10): 1-11.
Xie LG, Liao DX.A list of amoy mosquitoes with the description of a new species and a new variety[J].ActaEntomologicaSinica,1956,6(1): 123-128.[谢麟阁,廖定西.厦门蚊虫名录及一新种和变种的描述.昆虫学报,1956,6(1): 123-128]
Yang M, Chen HB. Research on 5 end sequences of 28s rRNA gene ofCulexvishnuicomplex[J].GuizhouScience, 2003, 21(1-2): 185-187.[杨明, 陈汉彬.杂鳞库蚊复组28S rRNA基因5′端序列研究[J].贵州科学, 2003, 21(1-2):185-187]
Molecular phylogeny ofCulexvishnuicomplex (Diptera: Culicidae) based on CO I and ITS sequences
ZHAO Wen-Jing, ZHANG Chun-Lin*, CHEN Han-Bin, ZHANG Jing, LIU Bin
(Department of Biology, Guizhou Medical University, Guiyang 550004, China)
Discussions about the classification status ofCulexannulusand the relationship ofC.vishnuicomplex were held with the Kimura-two-Parameter (K2P) distances based on CO I and phylogenetic trees based on CO I and ITS. The K2P distance betweenC.annulusandC.vishnuiwas 0.24%-0.72%, which supported “C.annulusis a synonym ofC.vishnui”.The interspecific K2P distance amongC.vishnui(containingC.annulus),C.pseudovishnuiandC.tritaeniorhynchuswere 4.41%-9.68%. From the view of the phylogenetic trees, each species respectively gathered and became sister group each other, which demonstrated again their being independent species of one another.C.annulusandC.vishnuibeing at the top of the phylogenetic trees indicated that they differentiated later, whileC.tritaeniorhynchusbeing at the root indicated that differentiated earlier.The intraspecific K2P distance ofC.tritaeniorhynchusmined in Japan was 0.48%-2.68%, and the interspecific K2P distances among those mined in Japan, China and India was 4.17%-6.76%.Furthermore, according to the phylogenetic trees,C.tritaeniorhynchusfrom Japan formed a branch themselves, then became the sister group with the branch formed by those from China and India. These results implied that JapaneseC.tritaeniorhynchushad the trend to differentiate at subspecies or even species level.
Culexvishnuicomplex; nucleotide sequences; K2P distance; phylogeny
贵州省科学技术基金项目(黔科合J字(2006)2077);贵州医科大学青年基金项目 (K2010-30)。
赵文静,女,硕士,主要从事昆虫分子学研究,E-mail: ellenzhwj@foxmail.com
Author for correspondence, E-mail: zcl@gmc.edu.cn
2015-10-18;接受日期Accepted:2015-12-16
Q963
A
1674-0858(2016)04-0821-10
猜你喜欢
植物研究(2023年5期)2023-09-09 08:01:22
河南师范大学学报(自然科学版)(2022年5期)2022-08-08 14:07:42
大自然探索(2021年9期)2021-11-07 21:12:58
工程力学(2021年6期)2021-07-06 07:01:40
云南畜牧兽医(2021年1期)2021-02-24 04:37:28
生物学通报(2019年7期)2019-07-08 06:04:02
天文爱好者(2016年7期)2016-12-20 09:30:16
传感技术学报(2016年1期)2016-03-22 02:26:59
杂草学报(2015年2期)2016-01-04 14:57:55
动物医学进展(2015年11期)2015-06-11 02:21:40
